Position Statement GIMBE

Evidence 2017;9(8): e1000170 doi: 10.4470/E1000170
Pubblicato: 17 ottobre 2017
Copyright: © 2016 Cartabellotta et al. Questo è un articolo open-access, distribuito con licenza Creative Commons Attribution, che ne consente l’utilizzo, la distribuzione e la riproduzione su qualsiasi supporto esclusivamente per fini non commerciali, a condizione di riportare sempre autore e citazione originale.

Il concetto di innovazione farmacologica, sin dall’incontro promosso dall’International Society of Drug Bulletins nel 2001 (1), continua ad essere oggetto di dibattito tra i differenti stakeholder del mondo farmaceutico: aziende produttrici, agenzie regolatorie, ricercatori, accademia, professionisti sanitari e pazienti. In particolare, troppo spesso il termine “innovativo” viene utilizzato per definire qualunque nuovo medicinale immesso in commercio, mentre sarebbe necessaria una definizione più rigorosa di tale termine.
L’innovazione farmacologica è fondamentale per migliorare la salute delle persone, ma il rischio di scambiare novità per innovazione è oggi molto elevato (2,3). Di conseguenza, agenzie di health technology assessment ed enti regolatori devono disporre di adeguati strumenti sia per valutare e classificare l’innovazione terapeutica, sia per assegnare la fascia di rimborsabilità e contrattare il prezzo con l’azienda, con il fine ultimo di ottenere il massimo ritorno in termini di salute dal denaro pubblico (4,5,6).
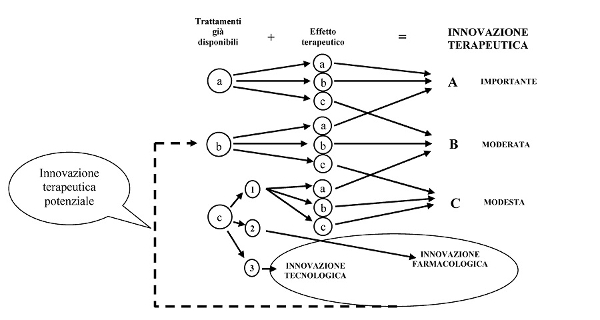
A tal fine, il 10 luglio 2007 la Commissione Tecnico Scientifica (CTS) dell’Agenzia Italiana per il Farmaco (AIFA) ha approvato un documento per valutare e classificare l’innovatività terapeutica di un nuovo farmaco tramite l’applicazione di uno specifico algoritmo che, grazie alla combinazione dei punteggi relativi ai trattamenti già disponibili e agli effetti terapeutici (7) (Appendice 1), permette di determinare il grado di innovazione terapeutica. Applicando quanto previsto da tale documento, l’AIFA ha periodicamente aggiornato l’elenco dei medicinali che possiedono il requisito dell’innovatività terapeutica “importante” ovvero “potenziale” (8), ai sensi dell’art.1 comma 1 dell’accordo 18 novembre 2010 (9).
Una svolta alle modalità di finanziamento dei farmaci innovativi è avvenuta con la Legge di Bilancio 2017 che stanzia € 500 milioni/anno sia per i farmaci innovativi, sia per i farmaci oncologici innovativi. L’erogazione di tali fondi è subordinata alla definizione da parte di AIFA, entro il 31 marzo 2017, di nuovi criteri per classificare i farmaci innovativi (Appendice 2).
Nel pieno rispetto dei tempi previsti dal comma 402 della Legge di Bilancio 2017, l’AIFA ha emanato la determina 519/2017 (10) – pubblicata in Gazzetta Ufficiale il 5 aprile 2017 – che approva i criteri per la classificazione dei farmaci innovativi, la procedura di valutazione e i criteri per la permanenza del requisito dell’innovatività, oltre che il modulo per la richiesta del riconoscimento dell’innovatività. Il 18 settembre 2017 l’AIFA, con la determina 1535/2017, ha aggiornato e sostituito la 519/2017 (11). Le principali modifiche relative a tale aggiornamento sono riportate nel box 1.
|
Box 1. Principali modifiche apportate dalla determina 1535/2017
|
1. Determina AIFA 1535/2017
Il modello di valutazione dell’innovatività del farmaco in relazione ad un’indicazione è unico per tutti i farmaci ma potrà prevedere, qualora necessario, l’utilizzo di ulteriori indicatori specifici.
1.1. Criteri per la valutazione dell’innovatività
Il modello proposto prevede un approccio multidimensionale che tiene conto di tre elementi:
- bisogno terapeutico
- valore terapeutico aggiunto
- qualità delle prove (evidenze scientifiche), ovvero la robustezza degli studi clinici.
1.1.1. Bisogno terapeutico
È condizionato dalla disponibilità di terapie per la patologia in oggetto e indica quanto l’introduzione di una nuova terapia sia necessaria per dare risposta alle esigenze terapeutiche di una popolazione di pazienti.
Ai fini del riconoscimento dell’innovatività del farmaco, il bisogno terapeutico può essere classificato in:
- Massimo: assenza di opzioni terapeutiche per la specifica indicazione.
- Importante: presenza di alternative terapeutiche per la specifica indicazione, ma che non producono impatto su esiti clinicamente rilevanti e validati per la patologia in oggetto.
- Moderato: presenza di alternative terapeutiche per la specifica indicazione con impatto limitato su esiti clinicamente rilevanti e/o con un profilo di sicurezza incerto o non del tutto soddisfacente.
- Scarso: presenza di una o più alternative terapeutiche per la specifica indicazione con impatto elevato su esiti clinicamente rilevanti e con un profilo di sicurezza favorevole.
- Assente: presenza di alternative terapeutiche per la specifica indicazione in grado di modificare la storia naturale della malattia e con un profilo di sicurezza favorevole.
1.1.2. Valore terapeutico aggiunto
È determinato dall’entità del beneficio clinico apportato dal nuovo farmaco rispetto alle alternative disponibili, se esistenti, su esiti riconosciuti come clinicamente rilevanti e validati per la patologia in oggetto. Per i farmaci oncologici la determina fornisce alcune indicazioni (box 2).
|
Box 2. Outcome in oncologia
|
Ai fini del riconoscimento dell’innovatività del farmaco in relazione all’indicazione specifica il valore terapeutico aggiunto può essere classificato in:
- Massimo: maggiore efficacia dimostrata su esiti clinicamente rilevanti rispetto alle alternative terapeutiche (se disponibili). Il farmaco è in grado di guarire la malattia o comunque di modificarne significativamente la storia naturale.
- Importante: maggiore efficacia dimostrata su esiti clinicamente rilevanti, o capacità di ridurre il rischio di complicazioni invalidanti o potenzialmente fatali, o migliore rapporto rischio/beneficio rispetto alle alternative, o capacità di evitare il ricorso a procedure cliniche ad alto rischio. Il farmaco modifica la storia naturale della malattia in una sottopopolazione di pazienti, o rappresenta comunque un vantaggio clinicamente rilevante, ad esempio in termini di qualità della vita e di intervallo libero dalla malattia, rispetto alle alternative terapeutiche disponibili.
- Moderato: maggiore efficacia di entità moderata o dimostrata in alcune sottopopolazioni di pazienti o su esiti surrogati, e con effetti limitati sulla qualità della vita. Per condizioni nelle quali sia ammissibile l’assenza di un comparatore, disponibilità di evidenze suggestive di migliore efficacia clinica e profilo R/B più favorevole rispetto alle alternative terapeutiche disponibili.
- Scarso: maggiore efficacia dimostrata su esiti clinicamente non rilevanti oppure di scarsa entità. Vantaggi minori (ad esempio via di somministrazione più favorevole) rispetto alle alternative terapeutiche disponibili.
- Assente: assenza di un beneficio clinico aggiuntivo rispetto alle alternative terapeutiche disponibili.
1.1.3. Qualità delle prove
La corretta valutazione del potenziale innovativo di un farmaco in relazione alla specifica indicazione dipende dalla qualità delle evidenze scientifiche a supporto della richiesta. Per valutare questo parametro l’AIFA ha adottato il metodo GRADE (Grading of Recommendations Assessment, Development and Evaluation) (12,13,14) che classifica la qualità delle evidenze in quattro categorie:
- Alta
- Moderata
- Bassa
- Molto bassa
1.2. Procedura di valutazione
La richiesta di riconoscimento del requisito di innovatività del farmaco in relazione alla singola indicazione deve essere sottomessa utilizzando l’apposito modulo predisposto da AIFA (15). L’AIFA, previo parere della CTS, può comunque valutare l’innovatività di un farmaco in relazione a una indicazione specifica in presenza di adeguate evidenze scientifiche, indipendentemente dalla richiesta di riconoscimento del requisito di innovatività da parte dell’azienda titolare.
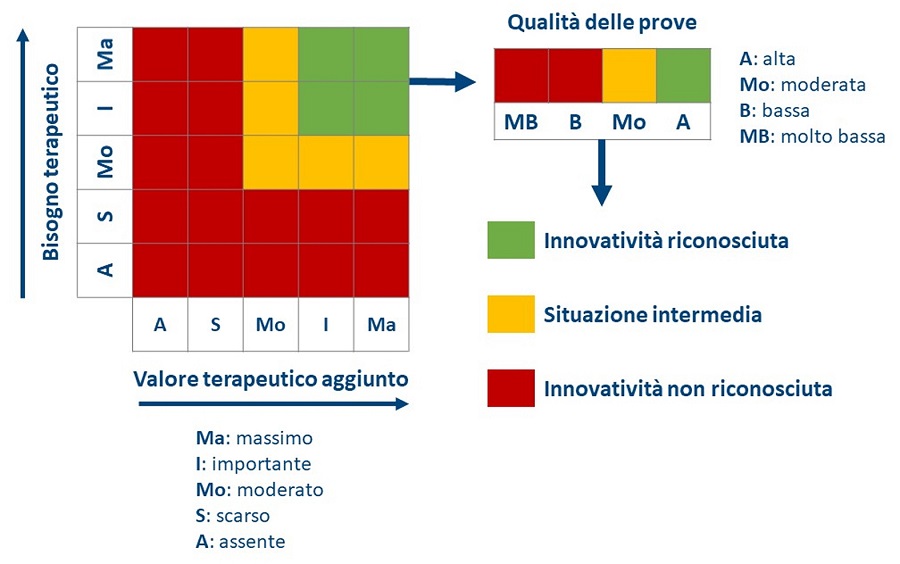
Il giudizio di innovatività viene formulato utilizzando la valutazione multidimensionale sopra descritta (bisogno terapeutico, valore terapeutico aggiunto, qualità delle prove), in relazione alla combinazione dei giudizi assegnati:
- Innovatività riconosciuta, in relazione alle singole indicazioni, se bisogno terapeutico e valore terapeutico aggiunto entrambi di livello “massimo” o “importante” e qualità delle prove “alta”.
- Innovatività non riconosciuta, se bisogno terapeutico e/o valore terapeutico aggiunto “scarso” o “assente”, oppure qualità delle prove “bassa” o “molto bassa”.
Le situazioni intermedie vengono valutate caso per caso, tenendo conto del peso relativo dei singoli elementi considerati.
Per i farmaci con indicazione per malattie rare, o con tassi di prevalenza ad esse assimilabili, la valutazione della qualità delle prove terrà conto della oggettiva difficoltà di condurre studi clinici gold standard e di adeguata potenza. In questi casi, pertanto, in presenza di un elevato bisogno terapeutico e di forti indicazioni di un beneficio terapeutico aggiunto, l’innovatività può essere attribuita anche sulla base di prove di qualità “bassa”.
La figura 1 rappresenta un tentativo di schematizzare le possibili combinazioni della valutazione multidimensionale per il riconoscimento della innovatività (17). Ovviamente, in assenza di ulteriori precisazioni da parte della determina, le “situazioni intermedie” potrebbero risultare da combinazioni differenti oltre quelle identificate. In ogni caso, se “innovatività riconosciuta” e “innovatività non riconosciuta” identificano due esiti del processo di valutazione, non è chiaro il destino delle “situazioni intermedie”, né le modalità con cui viene attribuita la “innovatività condizionata” (cfr. 1.3).
1.3. Esiti della valutazione
Al termine del processo, la CTS predispone una breve relazione, descrivendo le valutazioni su ciascuno dei tre ambiti considerati, e riporta l’esito della valutazione:
- riconoscimento dell’innovatività in relazione alla singola indicazione terapeutica, a cui saranno associati l’inserimento nel fondo dei farmaci innovativi, oppure nel Fondo dei farmaci innovativi oncologici, i benefici economici previsti dall’articolo 1, comma 403 della Legge di Bilancio 2017 e l’inserimento nei prontuari terapeutici regionali nei termini previsti dalla normativa vigente;
- riconoscimento dell’innovatività condizionata (o potenziale) in relazione alla singola indicazione terapeutica, che comporta solo l’inserimento nei prontuari terapeutici regionali nei termini previsti dalla normativa vigente;
- mancato riconoscimento dell’innovatività in relazione alla singola indicazione terapeutica.
La relazione viene trasmessa al richiedente, che può presentare controdeduzioni entro 10 giorni dalla comunicazione. L’esito finale e la relativa valutazione della CTS vengono rese pubbliche sul portale dell’AIFA contestualmente alla pubblicazione della determinazione di rimborsabilità e prezzo. Il richiedente, in fase di compilazione del modulo, può chiedere l’esclusione dalla pubblicazione di eventuali dati sensibili.
Come stabilito dall’articolo 1, comma 402, della Legge di Bilancio 2017, il riconoscimento dell’innovatività ed i benefici conseguenti hanno una durata massima di 36 mesi. La CTS attribuisce la durata dell’innovatività in relazione alla specifica indicazione al momento del riconoscimento del requisito. La permanenza del carattere di innovatività attribuito ad un farmaco può essere riconsiderata nel caso emergano evidenze che ne giustifichino la rivalutazione.
Nella rivalutazione di farmaci ad innovatività condizionata in relazione ad una specifica indicazione, la disponibilità di nuove evidenze valutate positivamente dall’AIFA, previo parere della CTS, può portare al riconoscimento dell’innovatività piena, con il conferimento dei benefici per il tempo residuo di durata prevista. In ogni caso, per i farmaci ad innovatività condizionata la sussistenza del requisito viene rivalutata decorsi 18 mesi dalla sua concessione, su istanza dell’azienda titolare o di ufficio.
In presenza di evidenze che smentiscano quelle che ne avevano giustificato il riconoscimento o ne ridimensionino l’effetto, l’innovatività del farmaco in relazione alla specifica indicazione può non essere confermata, e i benefici ad essa connessi decadono, con conseguente avvio di una nuova negoziazione del prezzo e delle condizioni di rimborsabilità.
In caso di autorizzazione di una nuova indicazione di un farmaco già riconosciuto come innovativo rispetto ad altra indicazione, la CTS può avviare, su richiesta o autonomamente, la valutazione del requisito dell’innovatività. In tal caso, la durata del beneficio non può superare i 36 mesi decorrenti dal riconoscimento dell’innovatività della nuova indicazione.
I benefici associati al riconoscimento dell’innovatività del farmaco in relazione alla specifica indicazione hanno la durata massima di 36 mesi per il farmaco first in class, mentre eventuali followers riconosciuti come innovativi per la stessa indicazione potranno beneficiarne per il periodo residuo.
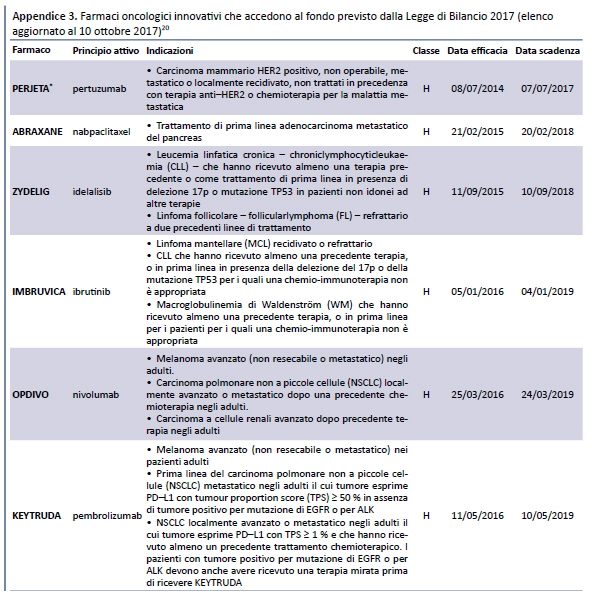
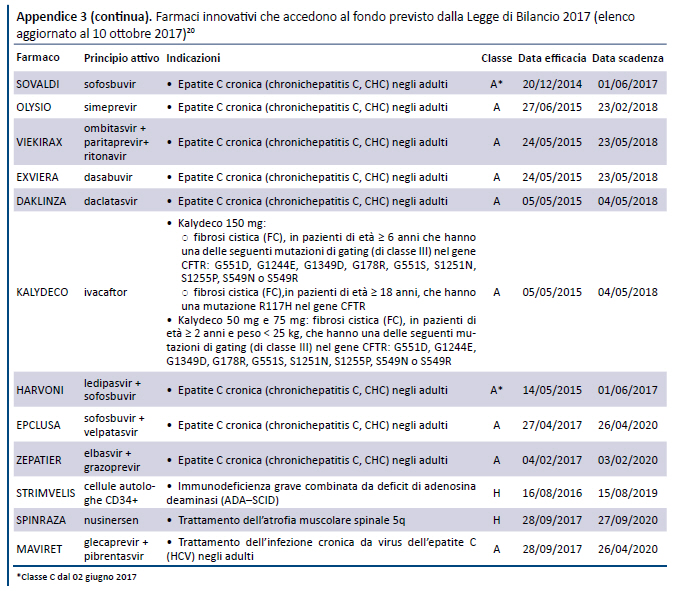
1.4. Elenchi dei farmaci che accedono ai fondi dei farmaci innovativi istituiti ai sensi della Legge di Bilancio 2017
Il 26 giugno 2017 l’AIFA ha reso disponibili gli elenchi dei farmaci innovativi che accedono ai fondi previsti dalla Legge di Bilancio 2017 (18): il primo relativo al “Fondo per il concorso al rimborso alle Regioni per l’acquisto dei medicinali innovativi” e il secondo al “Fondo per il concorso al rimborso alle Regioni per l’acquisto dei medicinali oncologici innovativi”. Secondo quanto riportato sul sito dall’AIFA, tali elenchi “sono stati elaborati tenendo conto di quanto disposto dal comma 402 della Legge di Bilancio 2017”, ovvero:
- secondo i pareri resi dalla CTS, a fronte dell’applicazione dei criteri definiti nella determinazione AIFA n. 519 del 31 marzo 2017, pubblicata nella GU n. 80 del 5 aprile 2017;
- considerando i farmaci innovativi e i farmaci oncologici innovativi già precedentemente identificati, per il periodo antecedente all’adozione della succitata determina.
I due elenchi, aggiornati periodicamente dall’AIFA sulla base dei pareri resi dalla CTS, hanno un formato standard che riporta: nome commerciale del farmaco, principio attivo, indicazioni, classe di rimborsabilità, data di efficacia (in considerazione della data di efficacia del relativo provvedimento di rimborsabilità), data di scadenza (in considerazione della data di efficacia e della durata del requisito di innovatività attribuito al farmaco/indicazione).
Tali elenchi sono stati successivamente aggiornati il 1 agosto 2017 (19) e il 10 ottobre 2017 (20) (appendice 3).
2. Analisi GIMBE
Nell’ambito delle attività dell’Osservatorio per la sostenibilità del Servizio Sanitario Nazionale (21) – a seguito delle richieste pervenute da società scientifiche, associazioni di pazienti e industria farmaceutica – la Fondazione GIMBE ha effettuato una valutazione metodologica indipendente della determina AIFA 1535/2017 e monitorato la sua applicazione rispetto al riconoscimento dell’innovatività e il conseguente accesso ai fondi assegnati dalla Legge di Bilancio 2017.
2.1. Analisi metodologica della determina AIFA
Considerata la possibilità di effettuare “integrazioni, aggiornamenti e revisioni in tempi successivi” prevista dalla determina, a seguito di un’analisi dei contenuti metodologici e delle procedure di assegnazione dell’innovatività, la Fondazione GIMBE ha rilevato alcune criticità e fornisce suggerimenti per la prossima revisione:
- Documento della CTS, 10 luglio 2007. I nuovi criteri definiti da AIFA per valutare l’innovazione terapeutica, ai sensi del comma 402 della Legge di Bilancio 2017, rappresentano un’evoluzione di tale documento che, tuttavia, non è mai stato formalmente rimosso né dalla determina 519/2017, né dal suo aggiornamento 1535/2017, che anzi lo richiamano nella premessa come “Documento della Commissione consultiva Tecnico-Scientifica concernente il Modello di valutazione dell’innovatività e la Procedura di valutazione finalizzata al riconoscimento del requisito di innovatività dei farmaci”. Anche se il documento della CTS non è mai stato pubblicato in Gazzetta Ufficiale e, pertanto, non è suscettibile di formale ritiro, è auspicabile che il prossimo aggiornamento della determina preveda il suo definitivo superamento, ad evitare il protrarsi dell’attuale sovrapposizione di due documenti tecnici con lo stesso obiettivo, ma che prevedono metodi differenti per valutare l’innovatività dei farmaci.
- Classificazione del bisogno terapeutico e del valore terapeutico aggiunto. La Fondazione GIMBE suggerisce di abolire il livello “moderato” per varie motivazioni:
- Il livello “moderato” non viene preso in considerazione nella procedura di valutazione (cfr 1.2), che fa riferimento solo ai livelli “massimo” o “importante” (ai fini del riconoscimento della innovatività) e “scarso” o “assente” (ai fini del non riconoscimento della innovatività).
- Le differenze con le categorie di valutazione (“importante” e “scarso”), adiacenti al livello “moderato” sono molto sfumate.
- L’utilizzo di scale con un numero dispari di item aumenta la probabilità di collocare il giudizio nella categoria mediana che, nel caso specifico, aumenta le “situazioni intermedie da valutare caso per caso”, e il rischio conseguente di discrezionalità e di possibili contenziosi.
- La scala a 5 item è disallineata rispetto a quella del GRADE (4 item), utilizzata per valutare la qualità delle prove.
- Outcome in oncologia. È opportuno dettagliare quali riferimenti vengono utilizzati nelle diverse tipologie di tumori, per “pesare” la variabile correlazione ben documentata in letteratura tra PFS, DSF ed altri end-point surrogati con la OS (22).
- Innovatività condizionata. È necessario definire esplicitamente i criteri con cui AIFA assegna l’innovatività condizionata rispetto alle 3 variabili previste dalla valutazione multidimensionale, al momento non disponibili nella determina 1535/2017.
- Malattie rare. Considerato che «nella valutazione della qualità delle prove […] in presenza di un elevato bisogno terapeutico e di forti indicazioni di un beneficio terapeutico aggiunto, l’innovatività può essere attribuita anche sulla base di prove di qualità “bassa”» è opportuno aggiungere anche “prove di qualità intermedia”.
- Innovatività riferita alla singola indicazione terapeutica. Nella determina 1535/2017 il concetto di innovatività viene sempre riferito alla singola indicazione terapeutica, tranne che nella valutazione del bisogno terapeutico. Ritenendo poco verosimile che possa trattarsi di una “svista”, è necessario precisare le ragioni per cui solo nella valutazione del bisogno terapeutico il concetto di innovatività non viene riferito alla singola indicazione terapeutica.
2.2. Elenchi dei farmaci innovativi
L’AIFA pubblica periodicamente gli elenchi dei farmaci innovativi sul proprio sito web, che, al momento della pubblicazione del presente Position Statement, riportata:
- 9 “elenchi aggiornati farmaci innovativi”, pubblicati in era pre-determina 519/2017 dal 23 dicembre 2010 al 21 dicembre 2016;
- 3 “elenchi dei farmaci che accedono ai fondi dei farmaci innovativi istituiti ai sensi della Legge di Bilancio 2017”, pubblicati in era post-determina 519/2017.
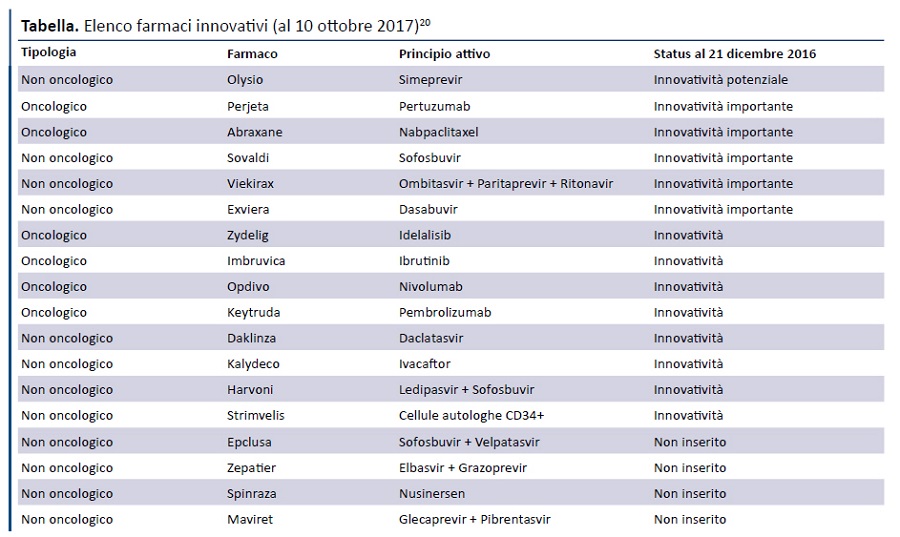
Come risulta dalla tabella, dei 18 farmaci innovativi inseriti nell’elenco del 10 ottobre 2017, 14 erano già presenti nell’ultimo elenco pre-determina 519/2017, pubblicato il 21 dicembre 2016 (23): di questi, 1 aveva ottenuto il requisito di “innovatività potenziale”, 5 di “innovatività importante”e 8 di “innovatività”.
Purtroppo, contrariamente a quanto riportato dalla determina 1535/2017 (“l’esito finale e la relativa valutazione della CTS saranno rese pubbliche sul portale dell’AIFA”), sul sito dell’AIFA non è disponibile alcuna documentazione che permetta di verificare l’applicazione del metodo multidimensionale per valutare l’innovatività dei farmaci che oggi accedono al fondo per l’innovazione, tenendo conto che:
- Dei 16 farmaci ai quali è stata riconosciuta l’innovatività:
- 14 erano già inseriti nell’ultimo elenco precedente alla pubblicazione della determina 519/2017, ma formalmente erano stati valutati con uno strumento differente (il documento della CTS del 10 luglio 2007).
- 4 sono stati inseriti successivamente, cui uno (sofosbuvir/velpatasvir) nell’intervallo tra il 21 dicembre e la pubblicazione della determina 519/2017 e gli altri (elbasvir/grazoprevir, nusinersen, glecaprevir + pibrentasvir) dopo la pubblicazione della predetta determina.
- Rispetto all’ultimo elenco dei farmaci innovativi pubblicato prima della determina 519/2017, 8 farmaci oggi non sono più considerati innovativi, ma è impossibile risalire alle motivazioni che, tramite l’applicazione della valutazione multidimensionale, hanno determinato il mancato riconoscimento.
Peraltro, la mancata disponibilità dei criteri con cui è stata applicata la determina 519/2017, rischia di mettere in contraddizione quanto previsto dal comma 402 della Legge di Bilancio (“Nelle more dell’adozione della determinazione di cui al presente comma e comunque entro e non oltre il 31 marzo 2017, i farmaci innovativi e i farmaci oncologici innovativi validi ai fini della presente procedura sono quelli già individuati dall’AIFA”) con quanto riportato sul sito dell’AIFA rispetto ai criteri utilizzati per assegnare l’innovatività: (“Gli elenchi sono stati elaborati tenendo conto di quanto disposto dal comma 402: secondo i pareri resi dalla Commissione consultiva tecnico-scientifica, a fronte dell’applicazione dei criteri definiti nella Determinazione AIFA n. 519 del 31 marzo 2017, pubblicata nella GU n. 80 del 5 aprile 2017; considerando i farmaci innovativi e i farmaci oncologici innovativi già precedentemente identificati, per il periodo antecedente all’adozione della succitata determina”).
Vero è che il comma 402 disponeva che sino alla pubblicazione della determina “i farmaci innovativi e i farmaci oncologici innovativi validi ai fini della presente procedura sono quelli già individuati dall’AIFA” e che AIFA doveva necessariamente considerare “i farmaci innovativi e i farmaci oncologici innovativi già precedentemente identificati, per il periodo antecedente all’adozione della succitata determina”; tuttavia, in mancanza di un report trasparente e completo sulla valutazione multidimensionale prevista dalla determina 1535/2017, è impossibile conoscere le motivazioni con cui, previa valutazione di bisogno terapeutico, valore terapeutico aggiunto e qualità delle prove, rispetto all’elenco pre-determina del 21 dicembre 2016, l’AIFA non ha confermato l’innovatività a 8 farmaci, ha confermato l’innovatività a 14 farmaci e ha assegnato l’innovatività a 4 nuovi farmaci (non presenti nell’elenco del 21 dicembre 2016). Infine, non è possibile verificare quanti e quali sono i farmaci per i quali le aziende farmaceutiche hanno presentato richiesta di innovatività senza averla riconosciuta.
Considerato che la determina AIFA per l’assegnazione dell’innovatività ha basi scientifiche e metodologiche ineccepibili, è indispensabile rendere pubblico – come peraltro previsto dalla stessa determina – il metodo con cui viene applicata, documentando il “peso” assegnato alle tre variabili (bisogno terapeutico, valore terapeutico aggiunto, qualità delle prove) che determinano l’esito finale del processo di valutazione (innovatività, innovatività condizionata, innovatività non riconosciuta).
La figura 2 riporta la modalità proposta dalla Fondazione GIMBE per un reporting esplicito del processo di valutazione, che dovrebbe essere pubblicato in occasione dell’aggiornamento periodico degli elenchi dei farmaci innovativi. Tale report dovrebbe essere reso disponibile per tutti i farmaci per i quali viene fatta richiesta di riconoscimento dell’innovatività, con l’opzione (in relazione alla volontà dell’azienda titolare) di anonimizzare il nome dei farmaci ai quali non viene riconosciuta l’innovatività.
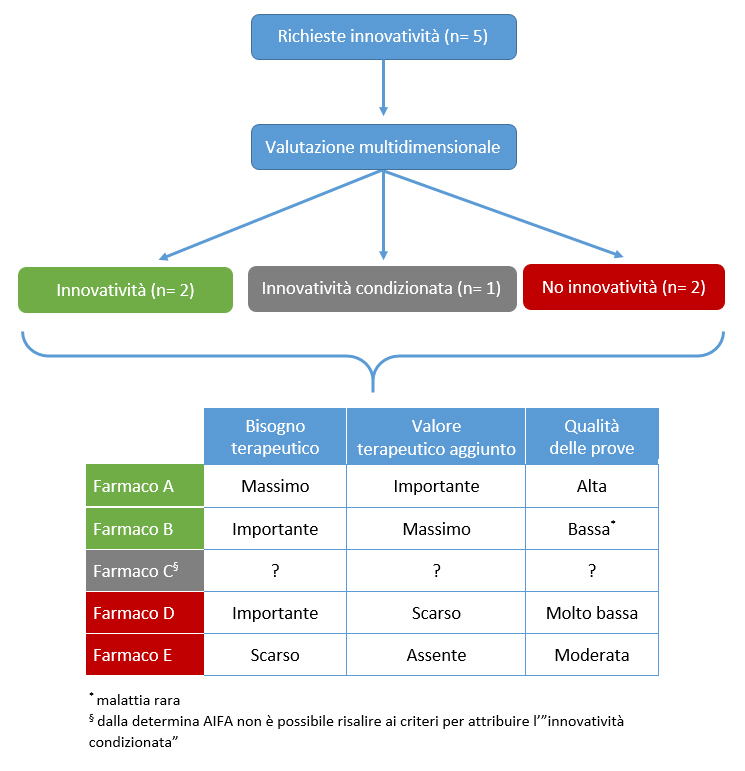
Conclusioni
Considerato che, senza un reporting trasparente e completo, il processo decisionale rimane implicito, auspichiamo che l’AIFA accolga le nostre proposte come elemento indispensabile di trasparenza sull’utilizzo del denaro pubblico, a beneficio dell’industria, di decisori, professionisti sanitari, pazienti, contribuenti tutti, oltre che di se stessa. Infatti, considerato che in tutti i processi decisionali sui farmaci l’AIFA mantiene sia la funzione di agenzia regolatoria, sia quella di health technology assessment solo processi decisionali espliciti e trasparenti possono spazzare via qualsiasi dubbio su questa “insolita” doppia veste di un ente che gestisce ogni anno quasi € 30 miliardi di spesa pubblica.
|
Appendice 1. Criteri definiti dal documento della Commissione Tecnico Scientifica del 10 luglio 2007, con relativo algoritmo (7) La classificazione preliminare di nuovi farmaci (o di nuove indicazioni terapeutiche di farmaci già noti) potenzialmente valutabili ai fini dell’innovazione terapeutica tiene conto innanzitutto delle malattie, suddivise in tre classi in ordine decrescente di importanza: Per attribuire il grado di innovazione terapeutica ai farmaci appartenenti a ciascuna delle tre classi, il documento AIFA considera due parametri: disponibilità di trattamenti ed entità dell’effetto terapeutico. Ciascun parametro è poi classificabile in tre fasce in ordine decrescente di importanza. In particolare rispetto alla disponibilità di trattamenti pre-esistenti: Al fine di assicurare un riconoscimento ai nuovi farmaci destinati al trattamento di malattie per le quali esistono già trattamenti disponibili, ma che possono presentare vantaggi rispetto alle terapie già esistenti, il punteggio C è stato ulteriormente suddiviso in tre sottogruppi, in ordine decrescente di importanza: L’attribuzione dell’entità dell’effetto terapeutico viene invece così classificata:
|
|
Appendice 2. LEGGE 11 dicembre 2016, n. 232. Art. 1. Disposizioni in merito di farmaci innovativi 400. A decorrere dal 1° gennaio 2017, nello stato di previsione del Ministero della salute è istituito un Fondo per il concorso al rimborso alle regioni per l’acquisto dei medicinali innovativi, con una dotazione di 500 milioni di euro annui [omissis]. 401. A decorrere dal 1°gennaio 2017, nello stato di previsione del Ministero della salute è istituito un Fondo per il concorso al rimborso alle Regioni per l’acquisto dei medicinali oncologici innovativi, con una dotazione di 500 milioni di euro annui, mediante utilizzo delle risorse del comma 393. 402. Per gli effetti di quanto previsto ai commi 400 e 401,con determinazione del direttore generale dell’Agenzia italiana del farmaco (AIFA), previo parere della Commissione consultiva tecnico-scientifica, da adottare entro il 31 marzo 2017, sono stabiliti i criteri per la classificazione dei farmaci innovativi e a innovatività condizionata e dei farmaci oncologici innovativi. Con la medesima determinazione sono definite le modalità per la valutazione degli effetti dei predetti farmaci ai fini della permanenza del requisito di innovatività e le modalità per la eventuale riduzione del prezzo di rimborso a carico del Servizio sanitario nazionale. Nelle more dell’adozione della determinazione di cui al presente comma e comunque entro e non oltre il 31 marzo 2017, i farmaci innovativi e i farmaci oncologici innovativi validi ai fini della presente procedura sono quelli già individuati dall’AIFA. 402-bis. I farmaci, ivi compresi quelli oncologici, per i quali è stato riconosciuto, da parte dell’AIFA, il possesso del requisito dell’innovatività condizionata, sono inseriti esclusivamentene i prontuari terapeutici regionali di cui all’articolo 10, commi 2 e 3, del decreto-legge 13 settembre 2012, n. 158, convertito, con modificazioni, dalla legge 8 novembre 2012, n. 189, e non accedono alle risorse di cui ai Fondi previsti ai commi 400 e 401 per un periodo massimo di diciotto mesi. Le risorse dei fondi di cui ai commi 400 e 401 non impiegate per le finalità ivi indicate confluiscono nella quota di finanziamento del fabbisogno sanitario nazionale standard cui concorre lo Stato ai sensi del comma 392 (16). 403. Il requisito di innovatività permane per un periodo massimodi 36 mesi. 404. I farmaci di cui al comma 402 sono soggetti a monitoraggio dei registri AIFA. 405. Le risorse dei fondi di cui ai commi 400 e 401 sono versate in favore delle Regioni in proporzione alla spesa sostenuta dalle regioni medesime per l’acquisto dei medicinali di cui ai citati commi 400 e 401, secondo le modalità individuate con apposito decreto del Ministro della Salute, di concerto con il Ministro dell’ Economia e delle Finanze, previa intesa in sede di Conferenza permanente per i rapporti tra lo Stato, le Regioni e le Province Autonome di Trento e di Bolzano. |