

Guidelines & Standards

Evidence 2012;4(7): e1000025 doi: 10.4470/E1000025
Pubblicato: 27 novembre 2012
Copyright: © 2012 Moher et al. Questo è un articolo open-access, distribuito con licenza Creative Commons Attribution, che ne consente l’utilizzo, la distribuzione e la riproduzione su qualsiasi supporto esclusivamente per fini non commerciali, a condizione di riportare sempre autore e citazione originale.
Vedi anche: CONSORT Statement 2010

Consistenti evidenze scientifiche dimostrano che la qualità del reporting dei trial controllati e randomizzati - randomized controlled trials (RCT) - non è ottimale. In assenza di un reporting chiaro e adeguato, i lettori non possono valutare l’affidabilità e la validità dei risultati di un trial, né sono in grado di identificare le informazioni necessarie alla produzione di revisioni sistematiche. Recenti studi metodologici dimostrano che un reporting e un disegno inadeguati si associano a stime distorte dell’efficacia del trattamento, compromettendo gravemente i RCT, considerati il gold standard per valutare l’efficacia degli interventi sanitari grazie alla loro capacità di minimizzare o evitare i bias.
Al fine di migliorare la qualità del reporting dei RCT un gruppo di ricercatori ed editori ha sviluppato il CONSORT (Consolidated Standards of Reporting Trials) Statement. Pubblicato nel 1996 e aggiornato nel 2001, il CONSORT Statement è costituito da una checklist e da un diagramma di flusso che gli autori possono utilizzare per il reporting di un RCT. Molte delle principali riviste mediche e i più importanti gruppi editoriali internazionali hanno adottato il CONSORT Statement che facilita l’approccio critico e l’interpretazione dei RCT.
Nel corso della revisione 2001 è emerso che una spiegazione ed elaborazione dei singoli item della checklist del CONSORT avrebbe agevolato ricercatori e altri utenti nella redazione o nella valutazione del reporting dei trial. Di conseguenza, la versione 2001 del CONSORT è stata integrata con un articolo di spiegazione ed elaborazione.
Nel gennaio 2007, in occasione di un meeting di esperti, il CONSORT è stato sottoposto a revisione e pubblicato come CONSORT Statement 2010, che ha migliorato la formulazione e la chiarezza della checklist precedente e include raccomandazioni su argomenti - come il bias di reporting selettivo degli outcome - di cui solo recentemente è stata presa consapevolezza.
Anche il presente documento di spiegazione ed elaborazione, finalizzato a migliorare la comprensione, l’utilizzo e la diffusione del CONSORT Statement, è stato sottoposto ad accurata revisione. Il presente documento illustra il background e il razionale scientifico di ciascun item - nuovo o aggiornato - del CONSORT 2010, fornendo esempi di un adeguato reporting, riferimenti bibliografici a studi sperimentali rilevanti e numerosi esempi di diagrammi di flusso.
Il CONSORT Statement 2010, la presente revisione del documento di spiegazione ed elaborazione e il sito web dedicato (www.consort-statement.org) costituiscono risorse indispensabili per migliorare il reporting dei RCT.
“Tutta la medicina dipende dalla trasparenza del reporting dei trial clinici” (1). Infatti, i trial controllati e randomizzati – randomized controlled trial (RCT) – ben disegnati e adeguatamente condotti forniscono le migliori prove di efficacia degli interventi sanitari, mentre quelli con metodologia inadeguata sono associati a bias e, in particolare, tendono a sovrastimare l’efficacia dei trattamenti (2-5). I risultati distorti di trial con disegno e reporting inadeguati possono determinare decisioni errate a tutti i livelli: dal trattamento del paziente individuale alle scelte nazionali di politica sanitaria.
Inoltre, la valutazione critica della qualità dei trial è possibile solo se il disegno, la conduzione e l’analisi dei RCT sono descritte in maniera completa e accurata. Lungi dall’essere trasparente, il reporting dei RCT è spesso incompleto (6-9), e comporta problemi conseguenti a una metodologia inadeguata (10-15).
Reporting incompleto e impreciso
Molte revisioni hanno documentato carenze nel reporting dei trial clinici: ad esempio, le informazioni sui metodi di assegnazione dei partecipanti a ciascun gruppo erano riportate solo nel 21% di 519 trial indicizzati in PubMed nel 2000 (16) e nel 34% dei 616 trial indicizzati nel 2006 (17). Analogamente, solo il 45% dei trial indicizzati in PubMed nel 200016 e il 53% nel 2006 (17) definivano un end-point primario, e solo il 27% nel 2000 e il 45% nel 2006 riportavano le metodologie utilizzate per stimare la dimensione del campione. Il reporting dei trial non solo è spesso incompleto, ma a volte è anche impreciso. Dei 119 trial in cui tutti i partecipanti erano analizzati secondo i gruppi di assegnazione originari (intention-to-treat analysis), 15 (13%) escludevano pazienti dall’analisi o non analizzavano tutti i pazienti nel gruppo di assegnazione (18). Numerose altre revisioni hanno riscontrato che un reporting inadeguato è più frequente nelle riviste specialistiche (16,19) e in quelle pubblicate in lingua non inglese (20,21).
Un’adeguata randomizzazione, riducendo il bias di selezione all’inizio del trial, è la componente fondamentale dei RCT di elevata qualità22. Una randomizzazione corretta richiede due step: la generazione di una sequenza di allocazione casuale e l’occultamento di tale sequenza ai ricercatori che arruolano i partecipanti (box 1) (2,23).
Box 1. Assegnazione dell’intervento: perché la randomizzazione è così importante? La metodologia utilizzata per assegnare i partecipanti a ciascun gruppo di intervento è un aspetto cruciale del disegno di un trial. L’assegnazione casuale è il metodo ideale, impiegato regolarmente con successo nei trial per oltre 50 anni (24). La randomizzazione ha tre vantaggi principali (25): in primo luogo, se applicata correttamente, elimina i bias di selezione, assicurando che tutti i fattori prognostici - sia noti che sconosciuti - si distribuiscano omogeneamente nel gruppo sperimentale e in quello di controllo. In assenza di randomizzazione, i confronti tra gli interventi possono risultare alterati, consapevolmente o meno, per la presenza del bias di selezione. In secondo luogo, l’assegnazione casuale consente di utilizzare la teoria della probabilità per esprimere la possibilità che ciascuna differenza di esito tra i gruppi di studio sia da attribuire all’efficacia del trattamento in studio (26). Infine, l’assegnazione casuale può in alcuni casi facilitare il blinding di ricercatori, partecipanti e valutatori degli esiti, grazie all’impiego di un placebo per ridurre i bias dopo l’assegnazione degli interventi (27). Di questi tre vantaggi, la prevenzione del bias di selezione all’inizio del trial è, indubbiamente, il più importante (27). Il successo della randomizzazione dipende da due aspetti strettamente correlati: un’adeguata generazione della sequenza di allocazione casuale e il suo occultamento sino all’assegnazione degli interventi (2,23). Un aspetto fondamentale è l’eventuale conoscenza o prevedibilità dell’assegnazione dei partecipanti ai gruppi di intervento da parte dei soggetti coinvolti nel trial (29). Il meccanismo di assegnazione degli interventi deve pertanto assicurare che chi arruola i partecipanti non conosca in anticipo a quale gruppo di intervento sarà assegnato il paziente successivo (occultamento della lista di randomizzazione) (2,23). Pertanto, se un’adeguata sequenza di allocazione consente di non prevedere le assegnazioni successive sulla base di quelle precedenti, un corretto occultamento della lista mantiene nascoste le assegnazioni successive. |
Purtroppo, nonostante il loro ruolo chiave, la descrizione delle metodologie utilizzate per l’assegnazione dei partecipanti agli interventi è generalmente inadeguata. Ad esempio, il 5% di 206 trial pubblicati in riviste di ostetricia e ginecologia erano in realtà studi non randomizzati (23). Questa stima è da considerarsi conservativa, poiché la maggior parte dei report attuali non descrive adeguatamente le metodologie di allocazione (20,23,30-33).
Come migliorare il reporting dei RCT: il CONSORT statement
DerSimonian et coll. hanno suggerito che “gli editori potrebbero migliorare sensibilmente il reporting di trial clinici, fornendo agli autori un elenco di item da riportare rigorosamente” (34). All’inizio degli anni ’90, due gruppi di editori, ricercatori e metodologi hanno pubblicato separatamente raccomandazioni per il reporting dei trial (35,36). A seguito di un invito di Rennie in un editoriale successivo, i due gruppi si sono incontrati sviluppando un set comune di indicazioni (37), dando vita al CONSORT (Consolidated Standards of Reporting Trials) Statement (38).
Il CONSORT Statement (o semplicemente CONSORT) comprende una checklist di item fondamentali che dovrebbero essere inclusi nel reporting dei RCT e un diagramma per documentare il flusso dei partecipanti nelle varie fasi di un trial. Pur essendo finalizzato a migliorare il reporting di trial a gruppi paralleli, numerosi item del CONSORT sono utili anche per altri disegni di trial: di non-inferiorità, di equivalenza, fattoriali, cluster e crossover. Per migliorare il reporting dei trial con questi disegni – così come il reporting di specifiche tipologie di dati (effetti avversi (42) ), di interventi (trattamenti non farmacologici (44), fitoterapia (44) ) e degli abstract (45), sono state pubblicate specifiche estensioni del CONSORT (39-41).
Il CONSORT ha l’obiettivo di fornire agli autori una guida per migliorare il reporting dei loro trial, rendendolo chiaro, completo e trasparente. Lettori, revisori ed editori possono utilizzare il CONSORT anche per la valutazione critica dei trial, ma è opportuno ricordare che il CONSORT non è stato elaborato con questa finalità. Molti item che, pur non definiti esplicitamente nel CONSORT, dovrebbero essere inclusi nel reporting di un trial: ad esempio le informazioni riguardanti l’approvazione del comitato etico, l’ottenimento del consenso informato da parte dei partecipanti, e, se rilevante, l’esistenza del comitato per la sicurezza dei dati e il monitoraggio. Inoltre, tutti gli altri aspetti di un trial che vengono menzionati devono essere adeguatamente riportati, come ad esempio i risultati di analisi di costo/efficacia (46-48).
Dalla sua prima pubblicazione nel 1996, il CONSORT è stato adottato da oltre 400 riviste (www.consort-statement.org) e promosso da diversi gruppi editoriali, come l’International Committee of Medical Journal Editors (49), determinando un miglioramento della qualità dei reporting dei trial (17,50,51). Tuttavia, il CONSORT è un’iniziativa in corso e il CONSORT Statement viene revisionato periodicamente (3): l’ultima revisione risale al 2001 (52-54). Da allora le evidenze scientifiche per informare il CONSORT sono notevolmente aumentate e dati empirici hanno evidenziato l’importanza di nuove criticità metodologiche, come il reporting selettivo degli outcome (55-57). Il gruppo CONSORT si è riunito in Canada nel gennaio 2007 per revisionare il CONSORT statement 2001 e il relativo documento di spiegazione ed elaborazione. La checklist revisionata è riportata nella tabella 1 e il diagramma di flusso, non revisionato, nella figura 1 (52-54).
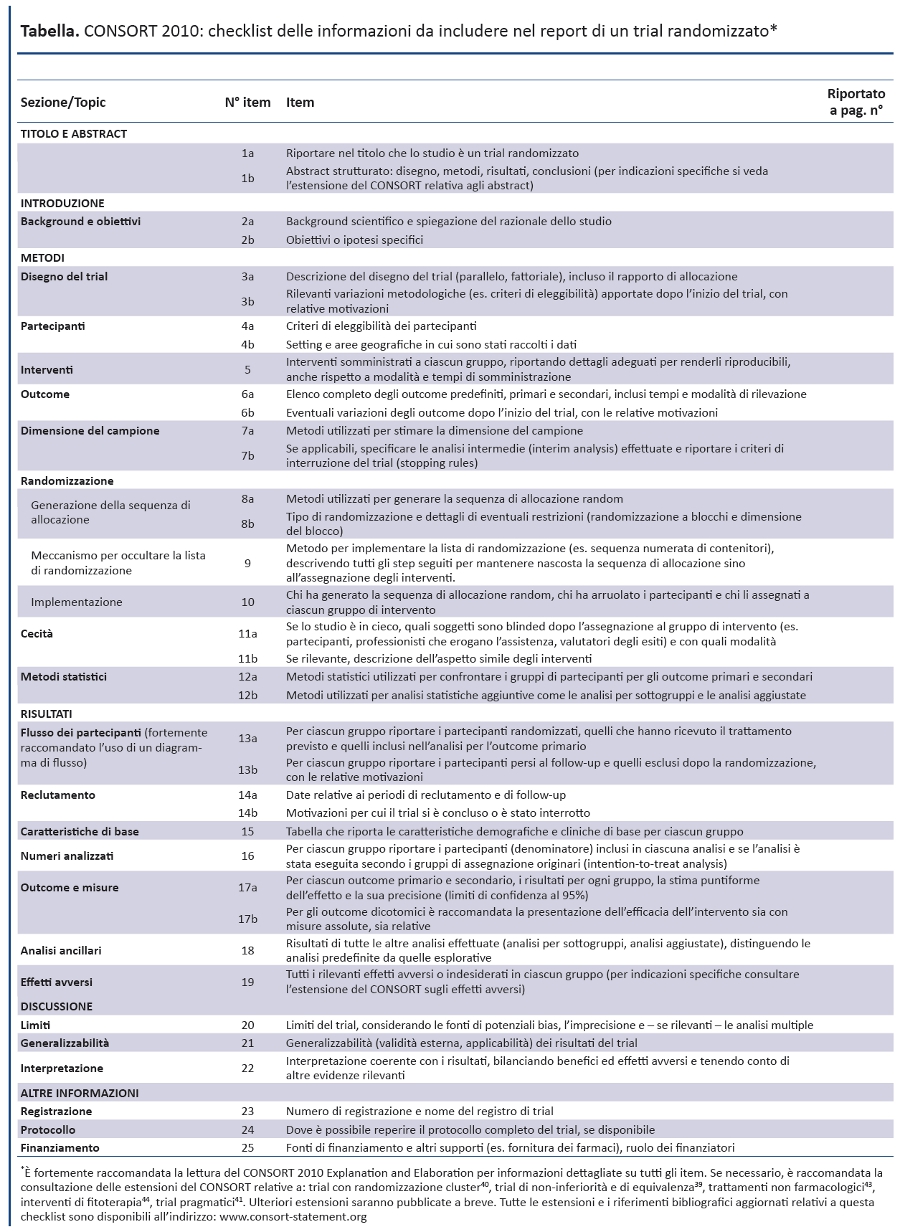
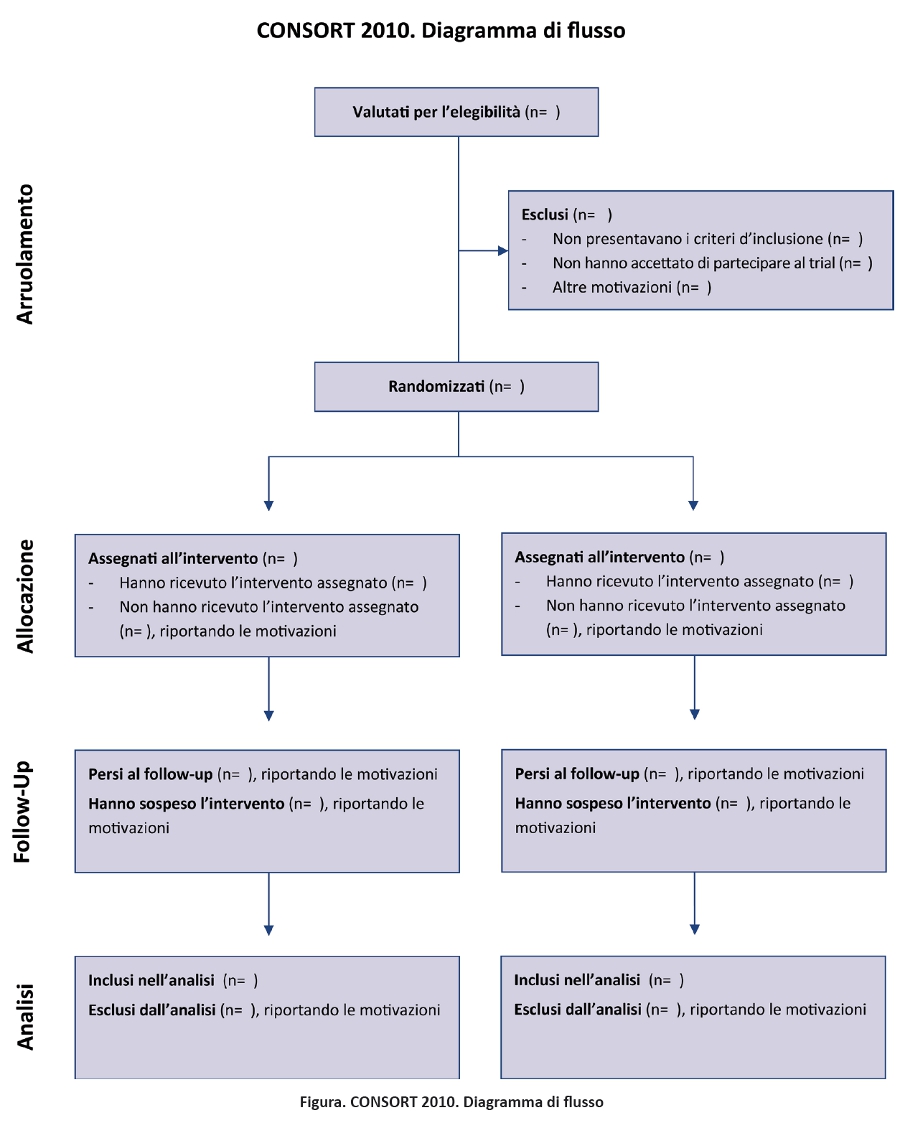
Il CONSORT Statement 2010: spiegazione ed elaborazione
Durante la revisione del CONSORT del 2001 è emerso che una spiegazione ed elaborazione dei singoli item avrebbero agevolato ricercatori e altri utenti nel reporting dei trial. Pertanto, insieme alla versione 2001 del CONSORT, è stato pubblicato un articolo di spiegazione ed elaborazione (58) che illustrava il background e il razionale scientifico di ogni item, riportando esempi pubblicati di reporting adeguato. Il razionale per la revisione dell’articolo di spiegazione ed elaborazione è simile a quello del CONSORT Statement. Descriviamo brevemente di seguito le principali considerazioni aggiunte e quelle eliminate della precedente versione dell’articolo di spiegazione ed elaborazione.
Le variazioni del CONSORT 2010 Spiegazione ed Elaborazione
Abbiamo apportato diverse modifiche sostanziali e alcune modifiche formali a questa versione del CONSORT 2010 Spiegazione ed Elaborazione (per i dettagli si veda la versione 2010 del CONSORT Statement (59) ). Alcune sono costituite da variazioni della checklist del CONSORT: ci sono tre item nuovi nella checklist del CONSORT 2010 – come l’item 24, che invita gli autori a indicare dove reperire il protocollo completo del trial. Abbiamo aggiornato alcune informazioni esistenti, riportando evidenze metodologiche più recenti e migliorato alcuni esempi. Abbiamo eliminato il glossario, ora disponibile sul sito web del CONSORT (www.consort-statement.org). Dove possibile, riportiamo ulteriori evidenze da studi empirici rilevanti. Molti libri eccellenti sui trial clinici offrono una discussione più ampia degli aspetti metodologici (60-62). Infine, per comodità, a volte utilizziamo la definizione “trattamenti” e “pazienti”, anche se non tutti gli interventi valutati nei RCT sono trattamenti e non tutti i partecipanti sono pazienti.
ITEM DELLA CHECKLIST
TITOLO E ABSTRACT
Item 1a. Riportare nel titolo che lo studio è un trial randomizzato.
Esempio. “Efficacia e sicurezza degli inalatori orali di nicotina per la disassuefazione al fumo: un trial clinico randomizzato in doppio cieco” (63).
Spiegazione. La capacità di identificare il report di un trial randomizzato in un database elettronico dipende in larga misura da come è stato indicizzato. È possibile che gli indicizzatori non classifichino un articolo come trial randomizzato se gli autori non riportano esplicitamente questa informazione (64). Per facilitare la corretta indicizzazione e l’identificazione di uno trial, gli autori dovrebbero utilizzare la parola “randomizzato” nel titolo, indicando che i partecipanti sono stati assegnati ai gruppi di confronto con modalità random.
Abstract strutturato: disegno, metodi, risultati, conclusioni (per indicazioni specifiche si veda l’estensione del CONSORT relativa agli abstract).
Per specifiche indicazioni si veda l’estensione del CONSORT relativa agli abstracts (45,65).
Spiegazione. E’ fondamentale che gli abstract siano chiari, trasparenti e sufficientemente dettagliati, perché spesso i lettori basano i loro giudizi esclusivamente su queste informazioni. Alcuni lettori utilizzano l’abstract come strumento di screening per decidere se leggere, o meno, l’articolo integrale. Inoltre, poiché non tutti i trial sono disponibili gratuitamente e non tutti i professionisti sanitari hanno accesso ai report completi dei trial, le decisioni cliniche vengono talora formulate sulla base delle informazioni contenute negli abstract (66).
Pertanto, l’abstract pubblicato su una rivista dovrebbe contenere informazioni adeguate e sufficienti da rappresentare una sintesi accurata delle metodologie e dei risultati del trial, secondo i vincoli editoriali e il formato della rivista.
Un abstract adeguatamente strutturato e ben redatto permette ai lettori di valutare rapidamente la rilevanza dei risultati e facilita il reperimento del trial nei database elettronici (67). L’abstract dovrebbe riflettere accuratamente i contenuti dell’articolo, senza includere informazioni che non compaiono nel testo integrale. Studi comparativi sull’accuratezza delle informazioni contenute negli abstract rispetto a quanto riportato nelle corrispondenti pubblicazioni integrali hanno rilevato dati incongruenti o mancanti rispetto all’articolo completo (68-71). Al contrario, omettere dall’abstract eventuali effetti avversi può indurre il lettore a interpretare erroneamente i risultati del trial (42,72).
Una recente estensione del CONSORT statement fornisce un elenco di item essenziali che gli autori devono includere pubblicando i risultati principali di un trial randomizzato in un abstract destinato ad una rivista o a un convegno (tabella 2) (45). Per il report di un trial randomizzato è fortemente raccomandato l’utilizzo di abstract strutturati, dove le informazioni sono organizzate in sezioni relative a disegno, conduzione, analisi e interpretazione (73). Alcuni studi hanno evidenziato che, rispetto agli abstract descrittivi, quelli strutturati hanno una qualità superiore (74,75) e permettono al lettore di identificare più facilmente le informazioni (76). In ogni caso, poiché numerose riviste hanno un proprio layout e impongono un limite di battute per gli abstract, non suggeriamo di modificare questi formati, ma ci limitiamo a raccomandare le informazioni da riportare.
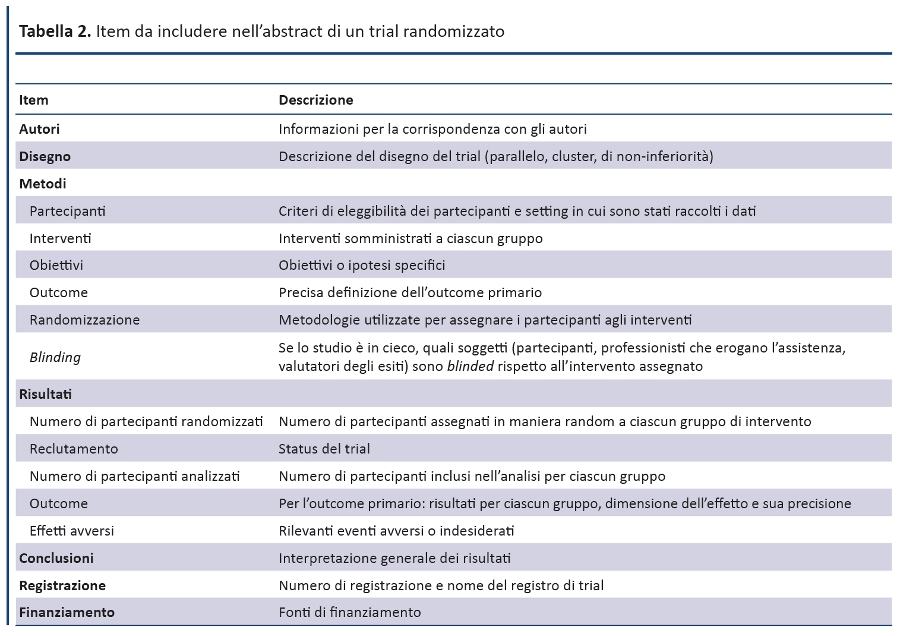
INTRODUZIONE
Item 2a. Background scientifico e spiegazione del razionale dello studio.
Esempio. “La chirurgia è il trattamento di prima scelta nei pazienti con carcinoma polmonare non a piccole cellule – non-small cell lung cancer (NSCLC) – di stadio I e II... Una meta-analisi sul NSCLC ha combinato i risultati di otto trial randomizzati che confrontavano l’intervento chirurgico rispetto alla combinazione chirurgia più chemioterapia adiuvante a base di cisplatino e ha mostrato un piccolo, ma non significativo (p = 0,08), beneficio assoluto di sopravvivenza di circa il 5% a 5 anni (dal 50% al 55%). Al momento della pianificazione del presente trial (metà degli anni ‘90), la chemioterapia adiuvante non era ancora il trattamento clinico standard... Il razionale scientifico per la chemioterapia neo-adiuvante è triplice: la regressione della neoplasia primaria potrebbe essere ottenuta in modo da semplificare o ridurre il successivo intervento chirurgico; micro-metastasi non identificate potrebbero essere considerate all’inizio del trattamento; si potrebbe inibire lo stimolo a neoplasie residue da parte di fattori di crescita rilasciati durante l’intervento chirurgico e la guarigione della ferita... il presente trial è stato quindi disegnato per confrontare, in pazienti con NSCLC resecabile, l’intervento chirurgico con tre cicli di chemioterapia a base di platino seguita dalla chirurgia in termini di sopravvivenza, qualità della vita, stadio di malattia, tasso di resecabilità, estensione della chirurgia, tempistiche e sedi delle recidive (77).”
Spiegazione. Di solito, l’introduzione è costituita da testo libero, dove gli autori spiegano il background e il razionale scientifico del trial e il suo schema generale. Può anche essere opportuno inserire nell’introduzione gli obiettivi del trial (item 2b). Il razionale può essere esplicativo (es., valutare la possibile influenza di un farmaco sulla funzione renale) o pragmatico (es., guidare la pratica clinica confrontando benefici ed effetti avversi di due interventi sanitari). Gli autori dovrebbero segnalare ogni evidenza di benefici ed effetti avversi degli interventi attivi inclusi in un trial e suggerire una spiegazione plausibile di come gli interventi dovrebbero funzionare, nel caso in cui non sia ovvio (78).
La Dichiarazione di Helsinki (79) sostiene che la ricerca biomedica che coinvolge soggetti umani dovrebbe essere basata su una conoscenza approfondita della letteratura scientifica, dal momento che non è etico esporre inutilmente gli esseri umani ai rischi della ricerca. Alcuni trial clinici sono risultati inutili perché il quesito valutato poteva essere risolto da una revisione sistematica della letteratura (80,81). Pertanto, la necessità di un nuovo trial deve essere giustificata nell’introduzione che, idealmente, dovrebbe includere un riferimento bibliografico a una revisione sistematica di trial simili già pubblicati, o una nota che specifichi che tali trial non sono ancora stati condotti (82).
Item 2b. Obiettivi o ipotesi specifici.
Esempio. “In questo studio abbiamo testato l’ipotesi che una gestione attiva del travaglio in donne nullipare dovrebbe: 1. Ridurre l’incidenza di taglio cesareo, 2. Ridurre l’incidenza di travaglio prolungato, 3. Non influenzare la soddisfazione materna durante l’esperienza del parto (83).”
Spiegazione. Gli obiettivi sono i quesiti a cui il trial intende dare una risposta, e spesso riguardano l’efficacia di uno specifico intervento terapeutico o preventivo. Le ipotesi sono i quesiti predefiniti che vengono valutati per poter raggiungere gli obiettivi. Le ipotesi sono più specifiche rispetto agli obiettivi e sottoposte a esplicita valutazione statistica; tuttavia, non sempre, obiettivi e ipotesi sono facilmente distinguibili. La maggior parte dei reporting di RCT fornisce adeguate informazioni relative a obiettivi e ipotesi del trial84.
METODI
Item 3a. Descrizione del disegno del trial (parallelo, fattoriale), incluso il rapporto di allocazione.
Esempio. “Studio multicentrico, stratificato (da 6 a 11 anni e da 12 a 17 anni di età, con randomizzazione non bilanciata [2:1]), in doppio cieco, controllato verso placebo, a gruppi paralleli condotto negli Stati Uniti (41 centri) (85).”
Spiegazione. Il termine “disegno” è spesso utilizzato per indicare tutti gli aspetti metodologici di pianificazione di un trial, ma può avere anche un’interpretazione più restrittiva. Molti aspetti specifici del disegno di un trial, come i dettagli di randomizzazione e cecità, sono trattati in altri item della checklist del CONSORT. In questa sezione sono presenti le informazioni sul tipo di trial, (es. a gruppi paralleli o fattoriale), il quadro concettuale (come la superiorità o la non-inferiorità), e altri aspetti correlati non analizzati in altri item della checklist.
Il CONSORT Statement si concentra principalmente sui trial con partecipanti randomizzati e assegnati individualmente a uno di due gruppi “paralleli”. In realtà, poco più del 50% dei trial pubblicati ha questo tipo di disegno (16). Tra i principali disegni alternativi ricordiamo i trial paralleli a bracci multipli, i trial con disegno crossover e fattoriali, i trial con randomizzazione cluster (40). Inoltre, se molti trial tendono, qualora esista, a dimostrare la superiorità di un nuovo intervento, altri ne valutano la non-inferiorità o l’equivalenza (39). È importante descrivere in modo chiaro questi aspetti dello studio, inclusa l’unità di randomizzazione (paziente, assistenza del medico di medicina generale, lesione), oltre a riportarli nell’abstract (item 1b).
Quando vengono utilizzati disegni di trial meno comuni, la scelta dovrebbe essere esplicitamente motivata, perché possono richiedere un campione di maggiori dimensioni, o analisi e interpretazioni più complesse.
Sebbene la maggior parte dei trial utilizzi la randomizzazione bilanciata (1:1 per due gruppi), si consiglia di esplicitare sempre il rapporto di allocazione. Per i trial farmacologici, può essere importante specificare anche la fase dello studio (I-IV).
Item 3b. Rilevanti variazioni metodologiche (es. criteri di eleggibilità) apportate dopo l’inizio del trial, con relative motivazioni.
Esempio. “I pazienti sono stati randomizzati e assegnati a uno dei sei gruppi paralleli, inizialmente secondo il rapporto 1:1:1:1:1:1, per ricevere uno dei cinque regimi di otamixaban [...] o un controllo attivo di eparina non frazionata [...] un comitato indipendente per il monitoraggio dei dati ha rivisto i dati relativi alla sicurezza del paziente non in cieco; non sono state eseguite analisi intermedie (interim analysis) per valutare l’efficacia. Durante il trial, il comitato ha raccomandato che il gruppo sottoposto alla dose più bassa di otamixaban (0,035 mg/kg/h) interrompesse il trattamento poiché la terapia anticoagulante risultava inadeguata. Sulla base di tale raccomandazione il protocollo è stato immediatamente modificato e i partecipanti sono stati successivamente randomizzati secondo il rapporto 2:2:2:2:1 rispettivamente ai 4 rimanenti gruppi di otamixaban e al controllo (86).”
Spiegazione. Se alcuni trial possono iniziare senza un protocollo stabilito (trial esplorativi), la maggior parte segue un protocollo che specifica nei dettagli le modalità di conduzione. Dal momento che è impossibile prevedere ogni eventuale cambiamento di situazioni, nel corso di un trial possono verificarsi deviazioni dal protocollo originale che richiederanno rilevanti variazioni metodologiche successive all’inizio del trial. Tali variazioni possono conseguire alla disponibilità di informazioni esterne derivanti da altri studi, a difficoltà finanziarie interne, oppure a un reclutamento inadeguato. Queste modifiche al protocollo dovrebbero essere apportate senza compromettere il blinding sugli outcome dei partecipanti. In alcuni trial, esiste un comitato indipendente di monitoraggio dei dati che può proporre variazioni del protocollo alla luce di dati non in cieco. Tali modifiche potrebbero influenzare i metodi di studio (es. variazioni di regimi di trattamento, criteri di eleggibilità, rapporto di randomizzazione o durata del follow-up) o la conduzione del trial (es. eliminazione di un centro con una scarsa qualità dei dati) (87).
Alcuni trial vengono progettati con un disegno “adattativo”. Non esiste una definizione universalmente accettata per questo disegno, ma in pratica lo si potrebbe definire “un disegno a più stadi che utilizza i dati raccolti per decidere come modificare gli aspetti dello studio, senza comprometterne validità e integrità” (88). Le modifiche, che solitamente riguardano la dimensione del campione e il numero dei bracci di trattamento, possono consentire decisioni più rapide e un più efficiente utilizzo delle risorse. Esistono, tuttavia, importanti aspetti etici, statistici e pratici da prendere in considerazione (89,90), e al fine di aiutare il lettore a interpretare i risultati, è essenziale riportare con estrema chiarezza se le modifiche apportate fanno parte del disegno dello studio o conseguono a mutate circostanze. Attualmente le variazioni metodologiche non vengono riportate in maniera adeguata: infatti, una revisione di confronti tra protocolli e rispettivi trial successivamente pubblicati ha evidenziato che circa la metà presenta inspiegabili discrepanze negli outcome primari (57), la randomizzazione, il blinding (91) e le analisi statistiche (92).
Item 4a. Criteri di eleggibilità dei partecipanti.
Esempio. “I partecipanti eleggibili erano gli adulti con infezione da HIV, di età ≥ 18 anni, che presentavano i criteri di eleggibilità per la terapia antiretrovirale secondo le linee guida nazionali del Malawi per il trattamento dell’HIV (stadio clinico III o IV OMS o qualsiasi stadio OMS con una conta di CD4 <250/mm3) e che all’inizio del trattamento avevano un BMI <18,5. I criteri di esclusione erano la gravidanza e l’allattamento, o partecipazione a un altro programma di nutrizione supplementare (93).”
Spiegazione. Per aiutare i lettori a interpretare lo studio, è necessario descrivere dettagliatamente i criteri di eleggibilità utilizzati per selezionare i partecipanti del trial. In particolare, una comprensione chiara di tali criteri è uno degli elementi indispensabili per stabilire a chi possono essere applicati i risultati di uno studio – cioè, la generalizzabilità del trial (applicabilità) e la rilevanza per la pratica clinica o la salute pubblica (item 21) (94). Altrettanto importante in questo senso è la descrizione del metodo di reclutamento, come l’invio selettivo o la partecipazione volontaria (es. attraverso la pubblicità). Dal momento che vengono applicati prima della randomizzazione, i criteri di eleggibilità non influenzano la validità interna di un trial, ma condizionano la sua validità esterna.
I criteri di eleggibilità tipici e ampiamente accettati dipendono dalla natura e dallo stadio della malattia oggetto di studio, mentre quelli di esclusione evitano l’arruolamento di partecipanti a rischio di eventi avversi dell’intervento in studio o per il rispetto di norme legali ed etiche. Il consenso informato dei partecipanti, ad esempio, viene tipicamente richiesto negli studi sperimentali. La distinzione tra criteri d’inclusione ed esclusione non è necessaria; lo stesso criterio può essere formulato per includere o escludere i partecipanti (95).
Nonostante la loro importanza, i criteri di eleggibilità spesso non vengono adeguatamente riportati. Ad esempio, otto trial pubblicati che hanno portato a segnalazioni cliniche da parte dei National Institutes of Health hanno descritto, in media, 31 criteri di eleggibilità nei loro protocolli, ma solo il 63% di tali criteri era riportato negli articoli pubblicati, e solo il 19% nelle segnalazioni cliniche (96). Carenze simili sono state riscontrate per i trial sull’HIV (97). Tra i 364 report di RCT in chirurgia, il 25% non ha indicato alcun criterio di eleggibilità (98).
Item 4b. Setting e aree geografiche in cui sono stati raccolti i dati.
Esempio. “Lo studio è stato eseguito presso il dipartimento di terapia antiretrovirale del Queen Elizabeth Central Hospital a Blantyre, Malawi, dal gennaio 2006 all’aprile 2007. Blantyre è la principale città commerciale del Malawi, con un 1.000.000 di abitanti e una prevalenza stimata di HIV del 27% negli adulti nel 2004 (93).”
Spiegazione. Oltre ai criteri di eleggibilità dei partecipanti (item 4a) e alla descrizione degli interventi (item 5), le informazioni sui setting e sulle aree geografiche sono fondamentali per valutare l’applicabilità di un trial. I partecipanti sono stati reclutati da setting di assistenza primaria, secondaria o terziaria o dalla comunità? Le istituzioni di assistenza sanitaria differiscono notevolmente in termini di organizzazione, esperienza e risorse e per quanto riguarda il rischio di base per la condizione in esame. Anche altri aspetti del setting (incluso il contesto sociale, economico e culturale e il clima) possono influenzare la validità esterna di uno studio.
Gli autori devono riportare il numero e la tipologia dei setting e descrivere i professionisti che erogano gli interventi sanitari. Devono inoltre riportare le aree geografiche in cui è stato condotto lo studio, inclusi il paese, se possibile la città, e l’ambiente circostante (es. la comunità, l’ambulatorio, l’ospedale o l’unità operativa). In particolare, dovrebbe essere chiaramente indicato se il trial è stato eseguito in uno o più centri (“trial multicentrici”). Questa descrizione dovrebbe fornire informazioni sufficienti per consentire ai lettori di valutare se i risultati dello studio possono essere rilevanti per il proprio setting assistenziale. Il contesto in cui viene condotto il trial può essere notevolmente diverso da quello in cui i suoi risultati saranno poi utilizzati per guidare la pratica clinica e le decisioni di politica sanitaria94,99. Gli autori devono fornire anche tutti gli altri dettagli relativi a setting e aree geografiche che potrebbero aver influenzato i risultati (es. problemi di viabilità e trasporti possono condizionare la partecipazione del paziente o determinare ritardi nella somministrazione degli interventi).
Item 5. Interventi somministrati a ciascun gruppo, riportando dettagli adeguati per renderli riproducibili, anche rispetto a modalità e tempi di somministrazione.
Esempi. “Nel trial POISE, i pazienti hanno ricevuto la prima dose del farmaco in studio (100 mg di metoprololo per via orale a rilascio prolungato) o del placebo 2-4 ore prima dell’intervento chirurgico. La somministrazione del farmaco richiedeva una frequenza cardiaca ≥ 50 bpm e una pressione sistolica ≥ 100 mmHg; questi parametri emodinamici sono stati monitorati prima di ogni somministrazione. Nelle prime 6 ore dopo l’intervento chirurgico, se in qualsiasi momento la frequenza cardiaca era ≥ 80 bpm e la pressione arteriosa sistolica ≥ 100 mmHg, i pazienti ricevevano la prima dose post-operatoria per via orale (100 mg di metoprololo a rilascio prolungato o placebo). Se il farmaco non era somministrato durante le prime 6 ore, i pazienti ricevevano la prima dose post-operatoria a 6 ore dall’intervento chirurgico. 12 ore dopo la prima dose post-operatoria, i pazienti iniziavano ad assumere 200 mg di metoprololo a rilascio prolungato per via orale o placebo, 1 volta/die per 30 giorni. Se la frequenza cardiaca del paziente era costantemente < 45 bpm o la pressione arteriosa sistolica scendeva sotto i 100 mmHg, il farmaco veniva sospeso sino ad un aumento della frequenza cardiaca o della pressione sistolica; il farmaco veniva, quindi, somministrato nuovamente al dosaggio di 100 mg/die. I pazienti in cui la frequenza cardiaca era costantemente 45-49 bpm e la pressione sistolica < 100 mmHg ritardavano l’assunzione del farmaco di 12 ore (100).”
“I pazienti erano randomizzati e assegnati a ricevere un tutore in neoprene su misura da indossare durante la notte oppure all’assistenza tradizionale. Il tutore, un’ortesi rigida raccomandata per uso esclusivamente notturno, ricopriva la base del pollice e il palmo, ma non il polso. I tutori erano stati realizzati da tre terapisti professionali esperti, che regolavano il tutore per ogni paziente in modo che il primo fotorecettore potesse essere aperto e il pollice posizionato dal lato opposto al primo dito lungo. I pazienti erano invitati a contattare il terapista se ritenevano necessaria una regolazione del tutore, in caso di aumento del dolore mentre indossavano il tutore, o di comparsa di effetti collaterali (es. erosione della pelle). Poiché in questa condizione non esiste nessun trattamento standard, i pazienti dei due gruppi (intervento e controllo) ricevevano cure tradizionali a discrezione del proprio medico di medicina generale o del reumatologo. Si è deciso di non utilizzare un placebo perché, secondo la nostra esperienza, nessun placebo per splintaggio è in grado di ottenere un blinding efficace dei pazienti (101).”
Spiegazione. Considerato che il medico che intende utilizzare l’intervento deve conoscerne esattamente le sue modalità di somministrazione nel trial, gli autori dovrebbero descrivere accuratamente ciascun intervento, inclusi quelli di controllo (102). Per un intervento farmacologico, le informazioni dovrebbero includere il nome del farmaco, la dose, le modalità di somministrazione (es. per via orale, endovenosa), le tempistiche e la durata della somministrazione, le condizioni in cui non è possibile erogare gli interventi e il regime di titolazione, se applicabile. Se il gruppo di controllo riceve “l’assistenza convenzionale” – usual care – è importante descrivere accuratamente da cosa è costituita. Se il gruppo sperimentale o quello di controllo riceve un intervento combinato, gli autori dovrebbero fornire sia una descrizione dettagliata di ciascun intervento, sia una spiegazione delle modalità con cui la combinazione degli interventi viene somministrata o interrotta, oltre ai fattori trigger per la loro introduzione, se applicabile.
Estensioni specifiche del CONSORT Statement riguardano il reporting di interventi non-farmacologici e fitoterapici ed evidenziano i dettagli specifici da prevedere (43,44), come il livello di competence professionale o i dettagli relativi alle modalità di standardizzazione degli interventi. Si raccomanda ai lettori di consultare le indicazioni per gli interventi non-farmacologici e fitoterapici, quando necessario.
Item 6a. Elenco completo degli outcome predefiniti, primari e secondari, inclusi tempi e modalità di rilevazione.
Esempio. “L’end-point primario relativo all’efficacia dell’intervento nella psoriasi era la percentuale di pazienti che raggiungeva un miglioramento del 75% dell’attività di malattia rispetto al valore di base dopo 12 settimane, secondo la misura del Psoriasis Area and Severity Index (PASI). Ulteriori analisi valutavano la variazione percentuale del punteggio PASI e il miglioramento delle lesioni psoriasiche (103).”
Spiegazione. Tutti i RCT valutano gli outcome (o end-point) per confrontare i risultati dei due gruppi di partecipanti. La maggior parte dei trial prevede diversi outcome, di cui alcuni hanno una maggiore rilevanza di altri. L’outcome primario è l’esito pre-definito considerato molto rilevante dai principali stakeholders (pazienti, policy-makers, medici, finanziatori) e viene utilizzato per stimare la dimensione del campione (item 7). La definizione di diversi outcome primari, anche se possibile, non è raccomandata perché può determinare problemi di interpretazione conseguenti alle analisi multiple (item 18 e 20). Gli outcome primari dovrebbero essere esplicitamente indicati come tali nel reporting di un RCT. Altri outcome di interesse sono quelli secondari (o accessori): gli outcome secondari possono essere diversi e spesso includono gli effetti avversi dell’intervento (item 19), che in realtà dovrebbero essere sempre considerati importanti, indipendentemente dal fatto che siano definiti primari o secondari.
Tutti gli outcome, primari e secondari, devono essere identificati e ben definiti nei dettagli per consentirne l’utilizzo ad altri ricercatori (102). Quando gli outcome vengono valutati in periodi diversi dopo la randomizzazione, gli autori dovrebbero indicare il time point pre-definito dell’outcome primario. Per molti interventi non farmacologici è utile specificare chi ha valutato gli esiti (es. se sono necessarie particolari competenze per farlo) e quanti erano i valutatori (43).
Qualora siano disponibili adeguate scale o linee guida, il loro utilizzo deve essere riportato (104,105), sia per migliorare la qualità delle misure, sia per consentire il confronto con studi simili (106). Ad esempio, la valutazione della qualità della vita può migliorare attraverso l’utilizzo di uno strumento validato (107). Gli autori dovrebbero sempre indicare la provenienza e le caratteristiche delle scale.
Oltre 70 outcome sono stati utilizzati in 196 RCT su farmaci anti-infiammatori non steroidei per l’artrite reumatoide (108), e 640 strumenti diversi sono stati utilizzati in 2.000 trial sulla schizofrenia, di cui 369 impiegati una sola volta (33). La valutazione di 149 di questi 2.000 trial ha dimostrato che l’utilizzo di scale non pubblicate in letteratura costituisce fonte di bias. Nei trial non farmacologici, un terzo delle definizioni di superiorità del trattamento, basato su scale non pubblicate, non sarebbe stato formulato se fosse stata utilizzata una scala pubblicata (109). Risultati analoghi sono stati riportati in altri campi (110,111). Solo il 45% di un gruppo di 519 RCT pubblicati nel 2000 specificava l’outcome primario16, analogamente al 53% di un gruppo simile di 614 RCT pubblicati nel 2006 (17).
Item 6b. Eventuali variazioni degli outcome dopo l’inizio del trial, con le relative motivazioni.
Esempio. “L’end-point primario originario era la mortalità per tutte le cause, ma, nel corso di un’analisi in cieco, il comitato di monitoraggio dei dati e della sicurezza ha riscontrato che la mortalità totale era inferiore a quella stimata e che lo studio non poteva essere completato con la dimensione del campione e la potenza definiti inizialmente. Il comitato direttivo ha pertanto deciso di utilizzare un duplice end-point primario: oltre alla mortalità per tutte le cause (end-point primario originario), la mortalità per tutte le cause insieme ai ricoveri per cause cardiovascolari (il primo end-point secondario predefinito) (112).”
Spiegazione. Esistono numerose motivazioni che richiedono di apportare variazioni al protocollo originale dello studio (item 24). Gli autori devono descrivere tutte le principali modifiche al protocollo, incluse quelle non previste ai criteri di inclusione, agli interventi, alle valutazioni, alla raccolta dei dati, ai metodi di analisi e agli outcome, informazioni che non sempre vengono riportate.
Come indicato in precedenza (item 6a), la maggior parte dei trial prende in considerazione diversi outcome, con il rischio di riportare i risultati solo per un sottogruppo selezionato (item 17); tale rischio dovrebbe essere prevenuto con la definizione preliminare e il reporting di outcome primari e secondari (item 6a). In alcuni trial, tuttavia, le circostanze richiedono una modifica delle modalità di valutazione di un outcome o addirittura, come nel precedente esempio, la modifica dell’outcome primario. Ad esempio, evidenze derivanti da altri trial o revisioni sistematiche possono suggerire che l’end-point potrebbe non essere appropriato; oppure il reclutamento dei partecipanti o l’incidenza dell’evento nel trial potrebbero essere inferiori al previsto (112). La variazione di un end-point sulla base di dati non in cieco è molto più problematica, anche se può essere specificata nel contesto di un trial con disegno adattativo88. Gli autori devono identificare e motivare tali modifiche, oltre che così riportare e spiegare eventuali variazioni agli outcome apportate dopo l’inizio del trial.
Un confronto tra protocolli e pubblicazioni di 102 trial randomizzati ha riscontrato che il 62% dei trial aveva almeno un outcome primario modificato, introdotto, oppure omesso rispetto al protocollo originale (55). Differenze negli outcome primari erano anche evidenti tra protocolli e pubblicazioni nel 40% di 48 trial finanziati dal Canadian Institutes of Health Research (113). Nessuno dei 150 report di trial successivamente pubblicati riportava, e tanto meno spiegava, le modifiche apportate al protocollo. Analoghi risultati provenienti da altri studi sono stati recentemente menzionati da una revisione sistematica di studi empirici che analizza i bias nel reporting degli outcome (57).
Item 7a. Metodi utilizzati per stimare la dimensione del campione.
Esempi. “Per rilevare una riduzione della degenza post-operatoria di 3 giorni (deviazione standard 5 giorni), in accordo con lo studio di Lobo et al. (17) con un livello di significatività del 5% e una potenza dell’80%, è stato stimato un campione di 50 pazienti per gruppo, considerando un’incidenza di abbandono del 10%. Per reclutare questo numero di pazienti è stato previsto un periodo di 12 mesi (114).”
“Sulla base di un’incidenza attesa dell’end-point primario composito del 11% dopo 2,25 anni nel gruppo placebo, abbiamo stimato necessari 950 eventi dell’end-point primario e un campione di 9.650 pazienti, con una potenza di 90% per rilevare una differenza significativa tra ivabradina e placebo, corrispondente a una riduzione relativa del 19% del rischio (errore a pari al 5%). Inizialmente abbiamo disegnato un trial event-driven, stabilendo di interromperlo una volta rilevati 950 end-point primari. Tuttavia, l’incidenza dell’end-point primario è stata superiore al previsto, forse per le caratteristiche di base dei pazienti reclutati, che presentavano un rischio più elevato rispetto a quello atteso (es. minore percentuale di pazienti classe NYHA I e incidenza più elevata di diabete e ipertensione). Abbiamo calcolato che nel momento in cui si fossero verificati 950 end-point primari, i pazienti inclusi più recentemente sarebbero stati trattati solo per circa 3 mesi. Di conseguenza, nel gennaio 2007, il comitato esecutivo ha deciso di modificare lo studio da event-driven a time-driven e di proseguire sino a 12 mesi il follow-up degli ultimi pazienti randomizzati. Questa variazione non ha modificato la durata di 3 anni prevista dello studio (115).”
Spiegazione. Per ragioni scientifiche ed etiche, la dimensione del campione di un trial deve essere accuratamente stimata, bilanciando valutazioni cliniche e statistiche. Idealmente, uno studio deve essere sufficientemente numeroso da avere un’elevata probabilità (potenza) di rilevare come statisticamente significativa una differenza clinicamente importante, se questa esiste realmente. La dimensione dell’effetto considerato importante è inversamente proporzionale alla dimensione del campione necessario per rilevarla: in altre parole per rilevare piccole differenze sono necessari campioni molto numerosi. Gli elementi per calcolare la dimensione del campione sono (1) l’incidenza degli outcome stimati in ciascun gruppo (che implica la differenza target clinicamente rilevante tra i gruppi di intervento); (2) l’errore a (tipo I); (3) la potenza statistica, oppure l’errore ß (tipo II); (4) per gli outcome continui, la deviazione standard (116). L’interazione di questi elementi e il loro reporting saranno diversi per i trial co
2.Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408-12.
3. Moher D. CONSORT: an evolving tool to help improve the quality of reports of randomized controlled trials. Consolidated Standards of Reporting Trials. JAMA 1998;279:1489-91.
4. Kjaergard LL, Villumsen J, Gluud C. Quality of randomised clinical trials affects estimates of intervention efficacy. 7th Cochrane Colloquium, Rome, Italy 1999.
5. Jüni P, Altman DG, Egger M. Systematic reviews in health care: Assessing the quality of controlled clinical trials. BMJ 2001;323:42-6.
6. Veldhuyzen van Zanten SJ, Cleary C, Talley NJ, Peterson TC, Nyren O, Bradley LA, et al. Drug treatment of functional dyspepsia: a systematic analysis of trial methodology with recommendations for design of future trials. Am J Gastroenterol 1996;91:660-73.
7. Talley NJ, Owen BK, Boyce P, Paterson K. Psychological treatments for irritable bowel syndrome: a critique of controlled treatment trials. Am J Gastroenterol1996;91:277-83.
8. Adetugbo K, Williams H. How well are randomized controlled trials reported in the dermatology literature? Arch Dermatol 2000;136:381-5
9. Kjaergard LL, Nikolova D, Gluud C. Randomized clinical trials in HEPATOLOGY: predictors of quality. Hepatology 1999;30:1134-8
10. Schor S, Karten I. Statistical evaluation of medical journal manuscripts. JAMA 1966;195:1123-8
11. Gore SM, Jones IG, Rytter EC. Misuse of statistical methods: critical assessment of articles in BMJ from January to March 1976. BMJ 1977;1:85-7.
12. Hall JC, Hill D, Watts JM. Misuse of statistical methods in the Australasian surgical literature. Aust N Z J Surg 1982;52:541-3.
13. Altman DG. Statistics in medical journals. Stat Med 1982;1:59-71.
14. Pocock SJ, Hughes MD, Lee RJ. Statistical problems in the reporting of clinical trials. A survey of three medical journals. N Engl J Med 1987;317:426-32
15. Altman DG. The scandal of poor medical research. BMJ 1994;308:283-4.
16. Chan AW, Altman DG. Epidemiology and reporting of randomised trials published in PubMed journals. Lancet 2005;365:1159-62.
17. Hopewell S, Dutton S, Yu LM, Chan AW, Altman DG. The quality of reports of randomised trials in 2000 and 2006: comparative study of articles indexed in PubMed. BMJ 2010;340:c723
18. Hollis S, Campbell F. What is meant by intention to treat analysis? Survey of published randomised controlled trials. BMJ 1999;319:670-4.
19. Lai TY, Wong VW, Lam RF, Cheng AC, Lam DS, Leung GM. Quality of reporting of key methodological items of randomized controlled trials in clinical ophthalmic journals. Ophthalmic Epidemiol 2007;14:390-8.
20. Moher D, Fortin P, Jadad AR, Jüni P, Klassen T, Le LJ, et al. Completeness of reporting of trials published in languages other than English: implications for conduct and reporting of systematic reviews. Lancet 1996;347:363-6.
21. Junker CA. Adherence to published standards of reporting: a comparison of placebo-controlled trials published in English or German. JAMA 1998;280:247-9.
22. Altman DG. Randomisation. BMJ 1991;302:1481-2.
23. Schulz KF, Chalmers I, Grimes DA, Altman DG. Assessing the quality of randomization from reports of controlled trials published in obstetrics and gynecology journals. JAMA 1994;272:125-8.
24. Streptomycin treatment of pulmonary tuberculosis: a Medical Research Council investigation. BMJ 1948;2:769-82.
25. Schulz KF. Randomized controlled trials. Clin Obstet Gynecol 1998;41:245-56.
26. Greenland S. Randomization, statistics, and causal inference. Epidemiology 1990;1:421-9.
27. Armitage P. The role of randomization in clinical trials. Stat Med 1982;1:345-52.
28. Kleijnen J, Gøtzsche PC, Kunz R, Oxman AD, Chalmers I. So what’s so special about randomisation. In: Maynard A, Chalmers I, eds. Non-random reflections on health services research. BMJ Books, 1997:93-106.
29. Chalmers I. Assembling comparison groups to assess the effects of health care. J R Soc Med 1997;90:379-86.
30. Nicolucci A, Grilli R, Alexanian AA, Apolone G, Torri V, Liberati A. Quality, evolution, and clinical implications of randomized, controlled trials on the treatment of lung cancer. A lost opportunity for meta-analysis. JAMA 1989;262:2101-7.
31. Ah-See KW, Molony NC. A qualitative assessment of randomized controlled trials in otolaryngology. J Laryngol Otol 1998;112:460-3.
32. Altman DG, Doré CJ. Randomisation and baseline comparisons in clinical trials. Lancet 1990;335:149-53.
33. Thornley B, Adams C. Content and quality of 2000 controlled trials in schizophrenia over 50 years. BMJ 1998;317:1181-4.
34. DerSimonian R, Charette LJ, McPeek B, Mosteller F. Reporting on methods in clinical trials. N Engl J Med 1982;306:1332-7.
35. A proposal for structured reporting of randomized controlled trials. The Standards of Reporting Trials Group. JAMA 1994;272:1926-31.
36. Call for comments on a proposal to improve reporting of clinical trials in the biomedical literature. Working Group on Recommendations for Reporting of Clinical Trials in the Biomedical Literature. Ann Intern Med 1994;121:894-5.
37. Rennie D. Reporting randomized controlled trials. An experiment and a call for responses from readers. JAMA 1995;273:1054-5.
38. Begg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, et al. Improving the quality of reporting of randomized controlled trials: the CONSORT statement. JAMA 1996;276:637-9.
39. Piaggio G, Elbourne DR, Altman DG, Pocock SJ, Evans SJ. Reporting of noninferiority and equivalence randomized trials: an extension of the CONSORT statement. JAMA 2006;295:1152-60.
40. Campbell MK, Elbourne DR, Altman DG. CONSORT statement: extension to cluster randomised trials. BMJ 2004;328:702-8.
41. Zwarenstein M, Treweek S, Gagnier JJ, Altman DG, Tunis S, Haynes B, et al. Improving the reporting of pragmatic trials: an extension of the CONSORT statement. BMJ 2008;337:a2390.
42. Ioannidis JP, Evans SJ, Gøtzsche PC, O’Neill RT, Altman DG, Schulz K, et al. Better reporting of harms in randomized trials: an extension of the CONSORT statement. Ann Intern Med 2004;141:781-8.
43. Boutron I, Moher D, Altman DG, Schulz KF, Ravaud P. Extending the CONSORT statement to randomized trials of nonpharmacologic treatment: explanation and elaboration. Ann Intern Med 2008;148:295-309.
44. Gagnier JJ, Boon H, Rochon P, Moher D, Barnes J, Bombardier C. Reporting randomized, controlled trials of herbal interventions: an elaborated CONSORT statement. Ann Intern Med 2006;144:364-7.
45. Hopewell S, Clarke M, Moher D, Wager E, Middleton P, Altman DG, et al. CONSORT for reporting randomized controlled trials in journal and conference abstracts: explanation and elaboration. PLoS Med 2008;5:e20.
46. Siegel JE, Weinstein MC, Russell LB, Gold MR. Recommendations for reporting cost-effectiveness analyses. Panel on Cost-Effectiveness in Health and Medicine. JAMA 1996;276:1339-41.
47. Drummond MF, Jefferson TO. Guidelines for authors and peer reviewers of economic submissions to the BMJ. The BMJ Economic Evaluation Working Party. BMJ 1996;313:275-83.
48. Lang TA, Secic M. How to report statistics in medicine. Annotated guidelines for authors, editors, and reviewers. ACP, 1997.
49. Davidoff F. News from the International Committee of Medical Journal Editors. Ann Intern Med 2000;133:229-31.
50. Plint AC, Moher D, Morrison A, Schulz K, Altman DG, Hill C, et al. Does the CONSORT checklist improve the quality of reports of randomised controlled trials? A systematic review. Med J Aust 2006;185:263-7.
51. Egger M, Jüni P, Bartlett C. Value of flow diagrams in reports of randomized controlled trials. JAMA 2001;285:1996-9.
52. Moher D, Schulz KF, Altman DG. The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomized trials. Ann Intern Med 2001;134:657-62.
53. Moher D, Schulz KF, Altman D. The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomized trials. JAMA 2001;285:1987-91.
54. Moher D, Schulz KF, Altman DG. The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomised trials. Lancet 2001;357:1191-4.
55. Chan AW, Hróbjartsson A, Haahr MT, Gøtzsche PC, Altman DG. Empirical evidence for selective reporting of outcomes in randomized trials: comparison of protocols to published articles. JAMA 2004;291:2457-65.
56. Al-Marzouki S, Roberts I, Evans S, Marshall T. Selective reporting in clinical trials: analysis of trial protocols accepted by the Lancet. Lancet 2008;372:201.
57. Dwan K, Altman DG, Arnaiz JA, Bloom J, Chan AW, Cronin E, et al. Systematic review of the empirical evidence of study publication bias and outcome reporting bias. PLoS ONE 2008;3:e3081.
58. Altman DG, Schulz KF, Moher D, Egger M, Davidoff F, Elbourne D, et al. The revised CONSORT statement for reporting randomized trials: explanation and elaboration. Ann Intern Med 2001;134:663-94.
59. Schulz KF, Altman DG, Moher D, for the CONSORT Group. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340:c332.
60. Pocock SJ. Clinical trials: a practical approach. John Wiley, 1983.
61. Meinert CL. Clinical trials: design, conduct and analysis. Oxford University Press, 1986.
62. Friedman LM, Furberg CD, DeMets DL. Fundamentals of clinical trials. 3rd ed. Springer, 1998.
63. Bolliger CT, Zellweger JP, Danielsson T, van Biljon X, Robidou A, Westin A, et al. Smoking reduction with oral nicotine inhalers: double blind, randomised clinical trial of efficacy and safety. BMJ 2000;321:329-33.
64. Dickersin K, Manheimer E, Wieland S, Robinson KA, Lefebvre C, McDonald S. Development of the Cochrane Collaboration’s CENTRAL Register of controlled clinical trials. Eval Health Prof 2002;25:38-64.
65. Hopewell S, Clarke M, Moher D, Wager E, Middleton P, Altman DG, et al. CONSORT for reporting randomised trials in journal and conference abstracts. Lancet 2008;371:281-3.
66. The impact of open access upon public health. PLoS Med 2006;3:e252.
67. Harbourt AM, Knecht LS, Humphreys BL. Structured abstracts in MEDLINE, 1989-1991. Bull Med Libr Assoc 1995;83:190-5.
68. Harris AH, Standard S, Brunning JL, Casey SL, Goldberg JH, Oliver L, et al. The accuracy of abstracts in psychology journals. J Psychol 2002;136:141-8.
69. Pitkin RM, Branagan MA, Burmeister LF. Accuracy of data in abstracts of published research articles. JAMA 1999;281:1110-1.
70. Ward LG, Kendrach MG, Price SO. Accuracy of abstracts for original research articles in pharmacy journals. Ann Pharmacother 2004;38:1173-7.
71. Gøtzsche PC. Believability of relative risks and odds ratios in abstracts: cross sectional study. BMJ 2006;333:231-4.
72. Ioannidis JP, Lau J. Completeness of safety reporting in randomized trials: an evaluation of 7 medical areas. JAMA 2001;285:437-43.
73. Haynes RB, Mulrow CD, Huth EJ, Altman DG, Gardner MJ. More informative abstracts revisited. Ann Intern Med 1990;113:69-76.
74. Taddio A, Pain T, Fassos FF, Boon H, Ilersich AL, Einarson TR. Quality of nonstructured and structured abstracts of original research articles in the British Medical Journal, the Canadian Medical Association Journal and the Journal of the American Medical Association. CMAJ 1994;150:1611-5.
75. Wager E, Middleton P. Technical editing of research reports in biomedical journals. Cochrane Database Syst Rev 2008;MR000002.
76. Hartley J, Sydes M, Blurton A. Obtaining information accurately and quickly: Are structured abstracts more efficient? J Inform Sci 1996;22:349-56.
77. Gilligan D, Nicolson M, Smith I, Groen H, Dalesio O, Goldstraw P, et al. Preoperative chemotherapy in patients with resectable non-small cell lung cancer: results of the MRC LU22/NVALT 2/EORTC 08012 multicentre randomised trial and update of systematic review. Lancet 2007;369:1929-37.
78. Sandler AD, Sutton KA, DeWeese J, Girardi MA, Sheppard V, Bodfish JW. Lack of benefit of a single dose of synthetic human secretin in the treatment of autism and pervasive developmental disorder. N Engl J Med 1999;341:1801-6.
79. World Medical Association. Declaration of Helsinki: ethical principle for medical research involving human subjects. 59th WMA General Assembly, Seoul 2008.
80. Lau J, Antman EM, Jimenez-Silva J, Kupelnick B, Mosteller F, Chalmers TC. Cumulative meta-analysis of therapeutic trials for myocardial infarction. N Engl J Med 1992;327:248-54.
81. Fergusson D, Glass KC, Hutton B, Shapiro S. Randomized controlled trials of aprotinin in cardiac surgery: could clinical equipoise have stopped the bleeding? Clin Trials 2005;2:218-29.
82. Savulescu J, Chalmers I, Blunt J. Are research ethics committees behaving unethically? Some suggestions for improving performance and accountability. BMJ 1996;313:1390-3.
83. Sadler LC, Davison T, McCowan LM. A randomised controlled trial and meta-analysis of active management of labour. BJOG 2000;107:909-15.
84. Bath FJ, Owen VE, Bath PM. Quality of full and final publications reporting acute stroke trials: a systematic review. Stroke 1998;29:2203-10.
85. Blumer JL, Findling RL, Shih WJ, Soubrane C, Reed MD. Controlled clinical trial of zolpidem for the treatment of insomnia associated with attention-deficit/hyperactivity disorder in children 6 to 17 years of age. Pediatrics 2009;123:e770-e776.
86. Sabatine MS, Antman EM, Widimsky P, Ebrahim IO, Kiss RG, Saaiman A, et al. Otamixaban for the treatment of patients with non-ST-elevation acute coronary syndromes (SEPIA-ACS1 TIMI 42): a randomised, double-blind, active-controlled, phase 2 trial. Lancet 2009;374:787-95.
87. Grant AM, Altman DG, Babiker AB, Campbell MK, Clemens FJ, Darbyshire JH, et al. Issues in data monitoring and interim analysis of trials. Health Technol Assess 2005;9:1-iv.
88. Gallo P, Krams M. PhRMA Working Group on adaptive designs, “White Paper.” Drug Information Journal 2006;40:421-82.
89. Brown CH, Ten Have TR, Jo B, Dagne G, Wyman PA, Muthen B, et al. Adaptive designs for randomized trials in public health. Annu Rev Public Health 2009;30:1-25.
90. Kelly PJ, Sooriyarachchi MR, Stallard N, Todd S. A practical comparison of group-sequential and adaptive designs. J Biopharm Stat 2005;15:719-38.
91. Pildal J, Chan AW, Hróbjartsson A, Forfang E, Altman DG, Gøtzsche PC. Comparison of descriptions of allocation concealment in trial protocols and the published reports: cohort study. BMJ 2005;330:1049.
92. Chan AW, Hróbjartsson A, Jørgensen KJ, Gøtzsche PC, Altman DG. Discrepancies in sample size calculations and data analyses reported in randomised trials: comparison of publications with protocols. BMJ 2008;337:a2299.
93. Ndekha MJ, van Oosterhout JJ, Zijlstra EE, Manary M, Saloojee H, Manary MJ. Supplementary feeding with either ready-to-use fortified spread or corn-soy blend in wasted adults starting antiretroviral therapy in Malawi: randomised, investigator blinded, controlled trial. BMJ 2009;338:1867-75.
94. Rothwell PM. External validity of randomised controlled trials: “to whom do the results of this trial apply?” Lancet 2005;365:82-93.
95. Fuks A, Weijer C, Freedman B, Shapiro S, Skrutkowska M, Riaz A. A study in contrasts: eligibility criteria in a twenty-year sample of NSABP and POG clinical trials. National Surgical Adjuvant Breast and Bowel Program. Pediatric Oncology Group. J Clin Epidemiol 1998;51:69-79.
96. Shapiro SH, Weijer C, Freedman B. Reporting the study populations of clinical trials. Clear transmission or static on the line? J Clin Epidemiol 2000;53:973-9.
97. Gandhi M, Ameli N, Bacchetti P, Sharp GB, French AL, Young M, et al. Eligibility criteria for HIV clinical trials and generalizability of results: the gap between published reports and study protocols. AIDS 2005;19:1885-96.
98. Hall JC, Mills B, Nguyen H, Hall JL. Methodologic standards in surgical trials. Surgery 1996;119:466-72.
99. Weiss NS, Koepsell TD, Psaty BM. Generalizability of the results of randomized trials. Arch Intern Med 2008;168:133-5.
100. Devereaux PJ, Yang H, Yusuf S, Guyatt G, Leslie K, Villar JC, et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet 2008;371:1839-47.
101. Rannou F, Dimet J, Boutron I, Baron G, Fayad F, Macé Y, et al. Splint for base-of-thumb osteoarthritis: a randomized trial. Ann Intern Med 2009;150:661-9.
102. Glasziou P, Meats E, Heneghan C, Shepperd S. What is missing from descriptions of treatment in trials and reviews? BMJ 2008;336:1472-4.
103. Mease PJ, Goffe BS, Metz J, VanderStoep A, Finck B, Burge DJ. Etanercept in the treatment of psoriatic arthritis and psoriasis: a randomised trial. Lancet 2000;356:385-90.
104. McDowell I, Newell C. Measuring health: a guide to rating scales and questionnaires. 3rd ed. New York: Oxford University Press, 2006.
105. Streiner D, Norman C. Health measurement scales: a practical guide to their development and use. 3rd ed. Oxford: Oxford University Press; 2003.
106. Clarke M. Standardising outcomes for clinical trials and systematic reviews. Trials 2007;8:39.
107. Sanders C, Egger M, Donovan J, Tallon D, Frankel S. Reporting on quality of life in randomised controlled trials: bibliographic study. BMJ 1998;317:1191-4.
108. Gøtzsche PC. Methodology and overt and hidden bias in reports of 196 double-blind trials of nonsteroidal antiinflammatory drugs in rheumatoid arthritis. Control Clin Trials 1989;10:31-56.
109. Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. Br J Psychiatry 2000;176:249-52.
110. Jadad AR, Boyle M, Cunnigham C, Kim M, Schachar R. Treatment of Attention-Deficit/Hyperactivity Disorder. Evidence Report/Technology Assessment No. 11. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Healthcare Research and Quality. AHQR publication no. 00-E005; 1999.
111. Schachter HM, Pham B, King J, Langford S, Moher D. The efficacy and safety of methylphenidate in attention deficit disorder: A systematic review and meta-analyis. Prepared for the Therapeutics Initiative, Vancouver, B.C., and the British Columbia Ministry for Children and Families, 2000.
112. Dargie HJ. Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: the CAPRICORN randomised trial. Lancet 2001;357:1385-90.
113. Chan AW, Krleza-Jeric K, Schmid I, Altman DG. Outcome reporting bias in randomized trials funded by the Canadian Institutes of Health Research. CMAJ 2004;171:735-40.
114. Vermeulen H, Hofland J, Legemate DA, Ubbink DT. Intravenous fluid restriction after major abdominal surgery: a randomized blinded clinical trial. Trials 2009;10:50.
115. Fox K, Ford I, Steg PG, Tendera M, Ferrari R. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a randomised, double-blind, placebo-controlled trial. Lancet 2008;372:807-16.
116. Campbell MJ, Julious SA, Altman DG. Estimating sample sizes for binary, ordered categorical, and continuous outcomes in two group comparisons. BMJ 1995;311:1145-8.
117. Guyatt GH, Mills EJ, Elbourne D. In the era of systematic reviews, does the size of an individual trial still matter. PLoS Med 2008;5:e4.
118. Schulz KF, Grimes DA. Sample size calculations in randomised trials: mandatory and mystical. Lancet 2005;365:1348-53.
119. Halpern SD, Karlawish JH, Berlin JA. The continuing unethical conduct of underpowered clinical trials. JAMA 2002;288:358-62.
120. Altman DG, Bland JM. Absence of evidence is not evidence of absence. BMJ 1995;311:485.
121. Moher D, Dulberg CS, Wells GA. Statistical power, sample size, and their reporting in randomized controlled trials. JAMA 1994;272:122-4.
122. Freiman JA, Chalmers TC, Smith H Jr, Kuebler RR. The importance of beta, the type II error and sample size in the design and interpretation of the randomized control trial. Survey of 71 “negative” trials. N Engl J Med 1978;299:690-4.
123. Charles P, Giraudeau B, Dechartres A, Baron G, Ravaud P. Reporting of sample size calculation in randomised controlled trials: review. BMJ 2009;338:b1732.
124. Yusuf S, Collins R, Peto R. Why do we need some large, simple randomized trials? Stat Med 1984;3:409-22.
125. Goodman SN, Berlin JA. The use of predicted confidence intervals when planning experiments and the misuse of power when interpreting results. Ann Intern Med 1994;121:200-6.
126. Galgiani JN, Catanzaro A, Cloud GA, Johnson RH, Williams PL, Mirels LF, et al. Comparison of oral fluconazole and itraconazole for progressive, nonmeningeal coccidioidomycosis. A randomized, double-blind trial. Mycoses Study Group. Ann Intern Med 2000;133:676-86.
127. Connolly SJ, Pogue J, Hart RG, Hohnloser SH, Pfeffer M, Chrolavicius S, et al. Effect of clopidogrel added to aspirin in patients with atrial fibrillation. N Engl J Med 2009;360:2066-78.
128. Geller NL, Pocock SJ. Interim analyses in randomized clinical trials: ramifications and guidelines for practitioners. Biometrics 1987;43:213-23.
129. Berry DA. Interim analyses in clinical trials: classical vs. Bayesian approaches. Stat Med 1985;4:521-6.
130. Pocock SJ. When to stop a clinical trial. BMJ 1992;305:235-40.
131. DeMets DL, Pocock SJ, Julian DG. The agonising negative trend in monitoring of clinical trials. Lancet 1999;354:1983-8.
132. Buyse M. Interim analyses, stopping rules and data monitoring in clinical trials in Europe. Stat Med 1993;12:509-20.
133. Sydes MR, Altman DG, Babiker AB, Parmar MK, Spiegelhalter DJ. Reported use of data monitoring committees in the main published reports of randomized controlled trials: a cross-sectional study. Clin Trials 2004;1:48-59.
134. Montori VM, Devereaux PJ, Adhikari NK, Burns KE, Eggert CH, Briel M, et al. Randomized trials stopped early for benefit: a systematic review. JAMA 2005;294:2203-9.
135. Coutinho IC, Ramos de Amorim MM, Katz L, Bandeira de Ferraz AA. Uterine exteriorization compared with in situ repair at cesarean delivery: a randomized controlled trial. Obstet Gynecol 2008;111:639-47.
136. Jüni P, Altman DG, Egger M. Assessing the quality of controlled clinical trials. In: Egger M, Davey Smith G, Altman DG, eds. Systematic reviews in health care: meta-analysis in context. BMJ Books, 2001.
137. Creinin MD, Meyn LA, Borgatta L, Barnhart K, Jensen J, Burke AE, et al. Multicenter comparison of the contraceptive ring and patch: a randomized controlled trial. Obstet Gynecol 2008;111:267-77.
138. Tate DF, Jackvony EH, Wing RR. Effects of internet behavioral counseling on weight loss in adults at risk for type 2 diabetes: a randomized trial. JAMA 2003;289:1833-6.
139. Lachin JM. Properties of simple randomization in clinical trials. Control Clin Trials 1988;9:312-26.
140. Peto R, Pike MC, Armitage P, Breslow NE, Cox DR, Howard SV, et al. Design and analysis of randomized clinical trials requiring prolonged observation of each patient. I. Introduction and design. Br J Cancer 1976;34:585-612.
141. Schulz KF, Grimes DA. The Lancet handbook of essential concepts in clinical research. Elsevier, 2006.
142. Altman DG, Bland JM. How to randomise. BMJ 1999;319:703-4.
143. Schulz KF. Subverting randomization in controlled trials. JAMA 1995;274:1456-8.
144. Enas GG, Enas NH, Spradlin CT, Wilson MG, Wiltse CG. Baseline comparability in clinical trials: prevention of poststudy anxiety. Drug Information Journal 1990;24:541-8.
145. Treasure T, MacRae KD. Minimisation: the platinum standard for trials? Randomisation doesn’t guarantee similarity of groups; minimisation does. BMJ 1998;317:362-3.
146. Sinei SK, Schulz KF, Lamptey PR, Grimes DA, Mati JK, Rosenthal SM, et al. Preventing IUCD-related pelvic infection: the efficacy of prophylactic doxycycline at insertion. Br J Obstet Gynaecol 1990;97:412-9.
147. Radford JA, Landorf KB, Buchbinder R, Cook C. Effectiveness of low-Dye taping for the short-term treatment of plantar heel pain: a randomised trial. BMC Musculoskelet Disord 2006;7:64.
148. Chalmers TC, Levin H, Sacks HS, Reitman D, Berrier J, Nagalingam R. Meta-analysis of clinical trials as a scientific discipline. I: Control of bias and comparison with large co-operative trials. Stat Med 1987;6:315-28.
149. Pocock SJ. Statistical aspects of clinical trial design. Statistician 1982;31:1-18.
150. Haag U. Technologies for automating randomized treatment assignment in clinical trials. Drug Information Journal 1998;32:11.
151. Piaggio G, Elbourne D, Schulz KF, Villar J, Pinol AP, Gülmezoglu AM. The reporting of methods for reducing and detecting bias: an example from the WHO Misoprostol Third Stage of Labour equivalence randomised controlled trial. BMC Med Res Methodol 2003;3:19.
152. Pildal J, Hróbjartsson A, Jórgensen KJ, Hilden J, Altman DG, Gøtzsche PC. Impact of allocation concealment on conclusions drawn from meta-analyses of randomized trials. Int J Epidemiol 2007;36:847-57.
153. Wood L, Egger M, Gluud LL, Schulz KF, Jüni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta-epidemiological study. BMJ 2008;336:601-5.
154. McCandlish R, Bowler U, van Asten H, Berridge G, Winter C, Sames L, et al. A randomised controlled trial of care of the perineum during second stage of normal labour. Br J Obstet Gynaecol 1998;105:1262-72.
155. Webster J, Clarke S, Paterson D, Hutton A, van Dyk S, Gale C, et al. Routine care of peripheral intravenous catheters versus clinically indicated replacement: randomised controlled trial. BMJ 2008;337:a339.
156. Smith SA, Shah ND, Bryant SC, Christianson TJ, Bjornsen SS, Giesler PD, et al. Chronic care model and shared care in diabetes: randomized trial of an electronic decision support system. Mayo Clin Proc 2008;83:747-57.
157. Sacks FM, Bray GA, Carey VJ, Smith SR, Ryan DH, Anton SD, et al. Comparison of weight-loss diets with different compositions of fat, protein, and carbohydrates. N Engl J Med 2009;360:859-73.
158. Kaptchuk TJ. Intentional ignorance: a history of blind assessment and placebo controls in medicine. Bull Hist Med 1998;72:389-433.
159. Guyatt GH, Pugsley SO, Sullivan MJ, Thompson PJ, Berman L, Jones NL, et al. Effect of encouragement on walking test performance. Thorax 1984;39:818-22.
160. Gøtzsche PC. Blinding during data analysis and writing of manuscripts. Control Clin Trials 1996;17:285-90.
161. Karlowski TR, Chalmers TC, Frenkel LD, Kapikian AZ, Lewis TL, Lynch JM. Ascorbic acid for the common cold. A prophylactic and therapeutic trial. JAMA 1975;231:1038-42.
162. Noseworthy JH, Ebers GC, Vandervoort MK, Farquhar RE, Yetisir E, Roberts R. The impact of blinding on the results of a randomized, placebo-controlled multiple sclerosis clinical trial. Neurology 1994;44:16-20.
163. Carley SD, Libetta C, Flavin B, Butler J, Tong N, Sammy I. An open prospective randomised trial to reduce the pain of blood glucose testing: ear versus thumb. BMJ 2000;321:20.
164. Schulz KF, Chalmers I, Altman DG. The landscape and lexicon of blinding in randomized trials. Ann Intern Med 2002;136:254-9.
165. Day SJ, Altman DG. Statistics notes: blinding in clinical trials and other studies. BMJ 2000;321:504.
166. Montori VM, Bhandari M, Devereaux PJ, Manns BJ, Ghali WA, Guyatt GH. In the dark: the reporting of blinding status in randomized controlled trials. J Clin Epidemiol 2002;55:787-90.
167. Cheng K, Smyth RL, Motley J, O’Hea U, Ashby D. Randomized controlled trials in cystic fibrosis (1966-1997) categorized by time, design, and intervention. Pediatr Pulmonol 2000;29:1-7.
168. Lang T. Masking or blinding? An unscientific survey of mostly medical journal editors on the great debate. Med Gen Med 2000;2:E25.
169. Devereaux PJ, Manns BJ, Ghali WA, Quan H, Lacchetti C, Montori VM, et al. Physician interpretations and textbook definitions of blinding terminology in randomized controlled trials. JAMA 2001;285:2000-3.
170. Haahr MT, Hróbjartsson A. Who is blinded in randomized clinical trials? A study of 200 trials and a survey of authors. Clin Trials 2006;3:360-5.
171. Meinert CL. Masked monitoring in clinical trials—blind stupidity? N Engl J Med 1998;338:1381-2.
172. Mills E, Prousky J, Raskin G, Gagnier J, Rachlis B, Montori VM, et al. The safety of over-the-counter niacin. A randomized placebo-controlled trial [ISRCTN18054903] . BMC Clin Pharmacol 2003;3:4.
173. Schulz KF, Grimes DA, Altman DG, Hayes RJ. Blinding and exclusions after allocation in randomised controlled trials: survey of published parallel group trials in obstetrics and gynaecology. BMJ 1996;312:742-4.
174. Fergusson D, Glass KC, Waring D, Shapiro S. Turning a blind eye: the success of blinding reported in a random sample of randomised, placebo controlled trials. BMJ 2004;328:432.
175. Sackett DL. Turning a blind eye: why we don’t test for blindness at the end of our trials. BMJ 2004;328:1136.
176. Astrup A, Rössner S, Van Gaal L, Rissanen A, Niskanen L, Al HM, et al. Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet 2009;374:1606-16.
177. Altman DG, Gore SM, Gardner MJ, Pocock SJ. Statistical guidelines for contributors to medical journals. In: Altman DG, Machin D, Bryant TN, Gardner MJ, eds. Statistics with confidence: confidence intervals and statistical guidelines. 2nd ed. BMJ Books, 2000:171-90.
178. Altman DG, Bland JM. Statistics notes. Units of analysis. BMJ 1997;314:1874.
179. Bolton S. Independence and statistical inference in clinical trial designs: a tutorial review. J Clin Pharmacol 1998;38:408-12.
180. Greenland S. Principles of multilevel modelling. Int J Epidemiol 2000;29:158-67.
181. Albert CM, Cook NR, Gaziano JM, Zaharris E, MacFadyen J, Danielson E, et al. Effect of folic acid and B vitamins on risk of cardiovascular events and total mortality among women at high risk for cardiovascular disease: a randomized trial. JAMA 2008;299:2027-36.
182. Matthews JN, Altman DG. Interaction 3: How to examine heterogeneity. BMJ 1996;313:862.
183. Assmann SF, Pocock SJ, Enos LE, Kasten LE. Subgroup analysis and other (mis)uses of baseline data in clinical trials. Lancet 2000;355:1064-9.
184. Matthews JN, Altman DG. Statistics notes. Interaction 2: Compare effect sizes not P values. BMJ 1996;313:808.
185. Oxman AD, Guyatt GH. A consumer’s guide to subgroup analyses. Ann Intern Med 1992;116:78-84.
186. Steyerberg EW, Bossuyt PM, Lee KL. Clinical trials in acute myocardial infarction: should we adjust for baseline characteristics? Am Heart J 2000;139:745-51.
187. Altman DG. Adjustment for covariate imbalance. In: Armitage P, Colton T, eds. Encyclopedia of biostatistics. John Wiley, 1998:1000-5.
188. Mullner M, Matthews H, Altman DG. Reporting on statistical methods to adjust for confounding: a cross-sectional survey. Ann Intern Med 2002;136:122-6.
189. Concato J, Feinstein AR, Holford TR. The risk of determining risk with multivariable models. Ann Intern Med 1993;118:201-10.
190. Bender R, Grouven U. Logistic regression models used in medical research are poorly presented. BMJ 1996;313:628.
191. Khan KS, Chien PF, Dwarakanath LS. Logistic regression models in obstetrics and gynecology literature. Obstet Gynecol 1999;93:1014-20.
192. Sackett DL, Gent M. Controversy in counting and attributing events in clinical trials. N Engl J Med 1979;301:1410-2.
193. May GS, DeMets DL, Friedman LM, Furberg C, Passamani E. The randomized clinical trial: bias in analysis. Circulation 1981;64:669-73.
194. Altman DG, Cuzick J, Peto J. More on zidovudine in asymptomatic HIV infection. N Engl J Med 1994;330:1758-9.
195. Meinert CL. Beyond CONSORT: need for improved reporting standards for clinical trials. Consolidated Standards of Reporting Trials. JAMA 1998;279:1487-9.
196. Grant AM, Wileman SM, Ramsay CR, Mowat NA, Krukowski ZH, Heading RC, et al. Minimal access surgery compared with medical management for chronic gastro-oesophageal reflux disease: UK collaborative randomised trial. BMJ 2008;337:a2664.
197. van Loon AJ, Mantingh A, Serlier EK, Kroon G, Mooyaart EL, Huisjes HJ. Randomised controlled trial of magnetic-resonance pelvimetry in breech presentation at term. Lancet 1997;350:1799-804.
198. Brown MJ, Palmer CR, Castaigne A, de Leeuw PW, Mancia G, Rosenthal T, et al. Morbidity and mortality in patients randomised to double-blind treatment with a long-acting calcium-channel blocker or diuretic in the International Nifedipine GITS study: Intervention as a Goal in Hypertension Treatment (INSIGHT). Lancet 2000;356:366-72.
199. LaCroix AZ, Ott SM, Ichikawa L, Scholes D, Barlow WE. Low-dose hydrochlorothiazide and preservation of bone mineral density in older adults. A randomized, double-blind, placebo-controlled trial. Ann Intern Med 2000;133:516-26.
200. Shuster JJ. Median follow-up in clinical trials. J Clin Oncol 1991;9:191-2.
201. Altman DG, de Stavola BL, Love SB, Stepniewska KA. Review of survival analyses published in cancer journals. Br J Cancer 1995;72:511-8.
202. Auvert B, Taljaard D, Lagarde E, Sobngwi-Tambekou J, Sitta R, Puren A. Randomized, controlled intervention trial of male circumcision for reduction of HIV infection risk: the ANRS 1265 Trial. PLoS Med 2005;2:e298.
203. Diggle L, Deeks J. Effect of needle length on incidence of local reactions to routine immunisation in infants aged 4 months: randomised controlled trial. BMJ 2000;321:931-3.
204. Pocock S, White I. Trials stopped early: too good to be true? Lancet 1999;353:943-4.
205. Hughes MD, Pocock SJ. Stopping rules and estimation problems in clinical trials. Stat Med 1988;7:1231-42.
206. Kiri A, Tonascia S, Meinert CL. Treatment effects monitoring committees and early stopping in large clinical trials. Clin Trials 2004;1:40-7.
207. Psaty BM, Rennie D. Stopping medical research to save money: a broken pact with researchers and patients. JAMA 2003;289:2128-31.
208. Temple R. FDA perspective on trials with interim efficacy evaluations. Stat Med 2006;25:3245-9.
209. Yusuf S, Teo K, Anderson C, Pogue J, Dyal L, Copland I, et al. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet 2008;372:1174-83.
210. Senn S. Base logic: tests of baseline balance in randomized clinical trials. Clin Res Regulatory Affairs 1995;12:171-82.
211. Altman DG. Comparability of randomised groups. Statistician 1985;34:125-36.
212. Heit JA, Elliott CG, Trowbridge AA, Morrey BF, Gent M, Hirsh J. Ardeparin sodium for extended out-of-hospital prophylaxis against venous thromboembolism after total hip or knee replacement. A randomized, double-blind, placebo-controlled trial. Ann Intern Med 2000;132:853-61.
213. Haderslev KV, Tjellesen L, Sorensen HA, Staun M. Alendronate increases lumbar spine bone mineral density in patients with Crohn’s disease. Gastroenterology 2000;119:639-46.
214. Fields WS, Maslenikov V, Meyer JS, Hass WK, Remington RD, Macdonald M. Joint study of extracranial arterial occlusion. V. Progress report of prognosis following surgery or nonsurgical treatment for transient cerebral ischemic attacks and cervical carotid artery lesions. JAMA 1970;211:1993-2003.
215. Lee YJ, Ellenberg JH, Hirtz DG, Nelson KB. Analysis of clinical trials by treatment actually received: is it really an option? Stat Med 1991;10:1595-605.
216. Lewis JA, Machin D. Intention to treat—who should use ITT? Br J Cancer 1993;68:647-50.
217. Lachin JL. Statistical considerations in the intent-to-treat principle. Control Clin Trials 2000;21:526.
218. Sheiner LB, Rubin DB. Intention-to-treat analysis and the goals of clinical trials. Clin Pharmacol Ther 1995;57:6-15.
219. Nagelkerke N, Fidler V, Bernsen R, Borgdorff M. Estimating treatment effects in randomized clinical trials in the presence of non-compliance. Stat Med 2000;19:1849-64.
220. Melander H, Ahlqvist-Rastad J, Meijer G, Beermann B. Evidence b(i)ased medicine--selective reporting from studies sponsored by pharmaceutical industry: review of studies in new drug applications. BMJ 2003;326:1171-3.
221. Gravel J, Opatrny L, Shapiro S. The intention-to-treat approach in randomized controlled trials: are authors saying what they do and doing what they say? Clin Trials 2007;4:350
222. Kruse RL, Alper BS, Reust C, Stevermer JJ, Shannon S, Williams RH. Intention-to-treat analysis: who is in? Who is out? J Fam Pract 2002;51:969-71.
223. Herman A, Botser IB, Tenenbaum S, Chechick A. Intention-to-treat analysis and accounting for missing data in orthopaedic randomized clinical trials. J Bone Joint Surg Am 2009;91:2137-43.
224. Ruiz-Canela M, Martinez-González MA, de Irala-Estévez J. Intention to treat analysis is related to methodological quality. BMJ 2000;320:1007-8.
225. Altman DG. Missing outcomes in randomised trials: addressing the dilemma. Open Med 2009;3:e21-3.
226. Wood AM, White IR, Thompson SG. Are missing outcome data adequately handled? A review of published randomized controlled trials in major medical journals. Clin Trials 2004;1:368-76.
227. Streiner DL. Missing data and the trouble with LOCF. Evid Based Ment Health 2008;11:3-5.
228. Molnar FJ, Hutton B, Fergusson D. Does analysis using “last observation carried forward” introduce bias in dementia research? CMAJ 2008;179:751-3.
229. Ware JH. Interpreting incomplete data in studies of diet and weight loss. N Engl J Med 2003;348:2136-7.
230. Streiner DL. The case of the missing data: methods of dealing with dropouts and other research vagaries. Can J Psychiatry 2002;47:68-75.
231. Lane P. Handling drop-out in longitudinal clinical trials: a comparison of the LOCF and MMRM approaches. Pharm Stat 2008;7:93-106.
232. Abraha I, Montedori A, Romagnoli C. Modified intention to treat: frequency, definition and implication for clinical trials [abstract]. Sao Paulo, Brazil: XV Cochrane Colloquium, 2007: 86-7.
233. Altman DG. Confidence intervals in practice. In: Altman DG, Machin D, Bryant TN, Gardner MJ, eds. Statistics with confidence. 2nd ed. BMJ Books, 2000:6-14.
234. van Linschoten R, van Middelkoop M, Berger MY, Heintjes EM, Verhaar JA, Willemsen SP, et al. Supervised exercise therapy versus usual care for patellofemoral pain syndrome: an open label randomised controlled trial. BMJ 2009;339:b4074.
235. Altman DG. Clinical trials and meta-analyses. In: Altman DG, Machin D, Bryant TN, Gardner MJ, eds. Statistics with confidence. 2nd ed. BMJ Books, 2000:120-38.
236. Uniform requirements for manuscripts submitted to biomedical journals. International Committee of Medical Journal Editors. Ann Intern Med 1997;126:36-47.
237. Gardner MJ, Altman DG. Confidence intervals rather than P values: estimation rather than hypothesis testing. BMJ 1986;292:746-50.
238. Bailar JC, III, Mosteller F. Guidelines for statistical reporting in articles for medical journals. Amplifications and explanations. Ann Intern Med 1988;108:266-73.
239. Bland JM. Quoting intermediate analyses can only mislead. BMJ 1997;314:1907-8.
240. Cook RJ, Sackett DL. The number needed to treat: a clinically useful measure of treatment effect. BMJ 1995;310:452-4.
241. Altman DG, Andersen PK. Calculating the number needed to treat for trials where the outcome is time to an event. BMJ 1999;319:1492-5.
242. The OSIRIS Collaborative Group. Early versus delayed neonatal administration of a synthetic surfactant—the judgment of OSIRIS (open study of infants at high risk of or with respiratory insufficiency—the role of surfactant). Lancet 1992;340:1363-9.
243. Sorensen L, Gyrd-Hansen D, Kristiansen IS, Nexøe J, Nielsen JB. Laypersons’ understanding of relative risk reductions: randomised cross-sectional study. BMC Med Inform Decis Mak 2008;8:31.
244. Bobbio M, Demichelis B, Giustetto G. Completeness of reporting trial results: effect on physicians’ willingness to prescribe. Lancet 1994;343:1209-11.
245. Naylor CD, Chen E, Strauss B. Measured enthusiasm: does the method of reporting trial results alter perceptions of therapeutic effectiveness? Ann Intern Med 1992;117:916-21.
246. Tukey JW. Some thoughts on clinical trials, especially problems of multiplicity. Science 1977;198:679-84.
247. Yusuf S, Wittes J, Probstfield J, Tyroler HA. Analysis and interpretation of treatment effects in subgroups of patients in randomized clinical trials. JAMA 1991;266:93-8.
248. Hahn S, Williamson PR, Hutton JL, Garner P, Flynn EV. Assessing the potential for bias in meta-analysis due to selective reporting of subgroup analyses within studies. Stat Med 2000;19:3325-36.
249. Bhandari M, Devereaux PJ, Li P, Mah D, Lim K, Schünemann HJ, et al. Misuse of baseline comparison tests and subgroup analyses in surgical trials. Clin Orthop Relat Res 2006;447:247-51.
250. Levin M, Quint PA, Goldstein B, Barton P, Bradley JS, Shemie SD, et al. Recombinant bactericidal/permeability-increasing protein (rBPI21) as adjunctive treatment for children with severe meningococcal sepsis: a randomised trial. rBPI21 Meningococcal Sepsis Study Group. Lancet 2000;356:961-7.
251. Scharf O, Colevas AD. Adverse event reporting in publications compared with sponsor database for cancer clinical trials. J Clin Oncol 2006;24:3933-8.
252. Pitrou I, Boutron I, Ahmad N, Ravaud P. Reporting of safety results in published reports of randomized controlled trials. Arch Intern Med 2009;169:1756-61.
253. Boden WE, O’Rourke RA, Teo KK, Hartigan PM, Maron DJ, Kostuk WJ, et al. Optimal medical therapy with or without PCI for stable coronary disease. N Engl J Med 2007;356:1503-16.
254. Horton R. The rhetoric of research. BMJ 1995;310:985-7.
255. Annals of Internal Medicine. Information for authors. Available at www.annals.org (accessed 15 Jan 2008).
256. Docherty M, Smith R. The case for structuring the discussion of scientific papers. BMJ 1999;318:1224-5.
257. Purcell GP, Donovan SL, Davidoff F. Changes to manuscripts during the editorial process: characterizing the evolution of a clinical paper. JAMA 1998;280:227-8.
258. Ioannidis JP. Limitations are not properly acknowledged in the scientific literature. J Clin Epidemiol 2007;60:324-9.
259. Kiviluoto T, Sirén J, Luukkonen P, Kivilaakso E. Randomised trial of laparoscopic versus open cholecystectomy for acute and gangrenous cholecystitis. Lancet 1998;351:321-5.
260. Hupperets MD, Verhagen EA, van Mechelen W. Effect of unsupervised home based proprioceptive training on recurrences of ankle sprain: randomised controlled trial. BMJ 2009;339:b2684.
261. Garber J, Clarke GN, Weersing VR, Beardslee WR, Brent DA, Gladstone TR, et al. Prevention of depression in at-risk adolescents: a randomized controlled trial. JAMA 2009;301:2215-24.
262. Campbell D. Factors relevant to the validity of experiments in social settings. Psychol Bull 1957;54:297-312.
263. Rothwell PM. Factors that can affect the external validity of randomised controlled trials. PLoS Clin Trials 2006;1:e9
264. King M, Nazareth I, Lampe F, Bower P, Chandler M, Morou M, et al. Conceptual framework and systematic review of the effects of participants’ and professionals’ preferences in randomised controlled trials. Health Technol Assess 2005;9:1-iv.
265. Djulbegovic B, Lacevic M, Cantor A, Fields KK, Bennett CL, Adams JR, et al. The uncertainty principle and industry-sponsored research. Lancet 2000;356:635-8.
266. Dans AL, Dans LF, Guyatt GH, Richardson S. Users’ guides to the medical literature: XIV. How to decide on the applicability of clinical trial results to your patient. Evidence-Based Medicine Working Group. JAMA 1998;279:545-9.
267. Smith GD, Egger M. Who benefits from medical interventions? BMJ 1994;308:72-4.
268. McAlister FA. Applying the results of systematic reviews at the bedside. In: Egger M, Davey Smith G, Altman DG, eds. Systematic reviews in health care: meta-analysis in context. BMJ Books, 2001.
269. Bartlett C, Doyal L, Ebrahim S, Davey P, Bachmann M, Egger M, et al. The causes and effects of socio-demographic exclusions from clinical trials. Health Technol Assess 2005;9:iii-x, 1.
270. Bonell C, Oakley A, Hargreaves J, Strange V, Rees R. Assessment of generalisability in trials of health interventions: suggested framework and systematic review. BMJ 2006;333:346-9.
271. Bornhöft G, Maxion-Bergemann S, Wolf U, Kienle GS, Michalsen A, Vollmar HC, et al. Checklist for the qualitative evaluation of clinical studies with particular focus on external validity and model validity. BMC Med Res Methodol 2006;6:56.
272. Laupacis A, Sackett DL, Roberts RS. An assessment of clinically useful measures of the consequences of treatment. N Engl J Med 1988;318:1728-33.
273. Altman DG. Confidence intervals for the number needed to treat. BMJ 1998;317:1309-12.
274. Fanaroff AA, Korones SB, Wright LL, Wright EC, Poland RL, Bauer CB, et al. A controlled trial of intravenous immune globulin to reduce nosocomial infections in very-low-birth-weight infants. National Institute of Child Health and Human Development Neonatal Research Network. N Engl J Med 1994;330:1107-13.
275. Randomised trial of intravenous atenolol among 16 027 cases of suspected acute myocardial infarction: ISIS-1. First International Study of Infarct Survival Collaborative Group. Lancet 1986;2:57-66.
276. Gøtzsche PC, Gjørup I, Bonnén H, Brahe NE, Becker U, Burcharth F. Somatostatin v placebo in bleeding oesophageal varices: randomised trial and meta-analysis. BMJ 1995;310:1495-8.
277. Clarke M, Hopewell S, Chalmers I. Reports of clinical trials should begin and end with up-to-date systematic reviews of other relevant evidence: a status report. J R Soc Med 2007;100:187-90.
278. Goodman SN. Toward evidence-based medical statistics. 1: The P value fallacy. Ann Intern Med 1999;130:995-1004.
279. Gøtzsche PC. Reference bias in reports of drug trials. BMJ 1987;295:654-6.
280. Papp K, Bissonnette R, Rosoph L, Wasel N, Lynde CW, Searles G, et al. Efficacy of ISA247 in plaque psoriasis: a randomised, multicentre, double-blind, placebo-controlled phase III study. Lancet 2008;371:1337-42.
281. Dickersin K. How important is publication bias? A synthesis of available data. AIDS Educ Prev 1997;9:15-21.
282. Song F, Eastwood AJ, Gilbody S, Duley L, Sutton AJ. Publication and related biases. Health Technol Assess 2000;4:1-115.
283. Williamson PR, Gamble C. Identification and impact of outcome selection bias in meta-analysis. Stat Med 2005;24:1547-61.
284. Tramèr MR, Reynolds DJ, Moore RA, McQuay HJ. Impact of covert duplicate publication on meta-analysis: a case study. BMJ 1997;315:635-40.
285. Simes RJ. Publication bias: the case for an international registry of clinical trials. J Clin Oncol 1986;4:1529-41.
286. Chalmers I. From optimism to disillusion about commitment to transparency in the medico-industrial complex. J R Soc Med 2006;99:337-41.
287. Tonks A. A clinical trials register for Europe. BMJ 2002;325:1314-5.
288. Dickersin K, Rennie D. Registering clinical trials. JAMA 2003;290:516-23.
289. Whittington CJ, Kendall T, Fonagy P, Cottrell D, Cotgrove A, Boddington E. Selective serotonin reuptake inhibitors in childhood depression: systematic review of published versus unpublished data. Lancet 2004;363:1341-5.
290. De Angelis CD, Drazen JM, Frizelle FA, Haug C, Hoey J, Horton R, et al. Is this clinical trial fully registered? A statement from the International Committee of Medical Journal Editors. Lancet 2005;365:1827-9.
291. Zarin DA, Ide NC, Tse T, Harlan WR, West JC, Lindberg DA. Issues in the registration of clinical trials. JAMA 2007;297:2112-20.
292. Hopewell S, Altman DG, Moher D, Schulz KF. Endorsement of the CONSORT Statement by high impact factor medical journals: a survey of journal editors and journal ‘instructions to authors’. Trials 2008;9:20.
293. Russell JA, Walley KR, Singer J, Gordon AC, Hébert PC, Cooper DJ, et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008;358:877-87.
294. Chan AW, Tetzlaff J, Altman D, Gøtzsche PC, Hróbjartsson A, Krleza-Jeric K, et al. The SPIRIT initiative: defining standard protocol items for randomised trials. Oral presentation at the 16th Cochrane Colloquium: Evidence in the era of globalisation; 2008 Oct 3-7; Freiburg, Germany [abstract]. Zeitschrift fur Evidenz, Fortbildung und Qualitat im Gesundheitswesen 2008;102:27.
295. Gregson S, Adamson S, Papaya S, Mundondo J, Nyamukapa CA, Mason PR, et al. Impact and process evaluation of integrated community and clinic-based HIV-1 control: a cluster-randomised trial in eastern Zimbabwe. PLoS Med 2007;4:e102.
296. Shaddy RE, Boucek MM, Hsu DT, Boucek RJ, Canter CE, Mahony L, et al. Carvedilol for children and adolescents with heart failure: a randomized controlled trial. JAMA 2007;298:1171-9.
297. Lexchin J, Bero LA, Djulbegovic B, Clark O. Pharmaceutical industry sponsorship and research outcome and quality: systematic review. BMJ 2003;326:1167-70.
298. Kjaergard LL, Als-Nielsen B. Association between competing interests and authors’ conclusions: epidemiological study of randomised clinical trials published in the BMJ. BMJ 2002;325:249.
299. Bero L, Oostvogel F, Bacchetti P, Lee K. Factors associated with findings of published trials of drug-drug comparisons: why some statins appear more efficacious than others. PLoS Med 2007;4:e184.
300. Sismondo S. Pharmaceutical company funding and its consequences: a qualitative systematic review. Contemp Clin Trials 2008;29:109-13.
301. Als-Nielsen B, Chen W, Gluud C, Kjaergard LL. Association of funding and conclusions in randomized drug trials: a reflection of treatment effect or adverse events? JAMA 2003;290:921-8.
302. Ross JS, Hill KP, Egilman DS, Krumholz HM. Guest authorship and ghostwriting in publications related to rofecoxib: a case study of industry documents from rofecoxib litigation. JAMA 2008;299:1800-12.
303. McAlister FA, Straus SE, Sackett DL, Altman DG. Analysis and reporting of factorial trials: a systematic review. JAMA 2003;289:2545-53.
304. Senn S. Crossover trials in clinical research. 2nd ed. Wiley, 2002.
305. Deeks JJ, Dinnes J, D’Amico R, Sowden AJ, Sakarovitch C, Song F, et al. Evaluating non-randomised intervention studies. Health Technol Assess 2003;7:iii-173.
306. Kunz R, Vist G, Oxman AD. Randomisation to protect against selection bias in healthcare trials. Cochrane Database Syst Rev 2007;MR000012.
307. Collins R, MacMahon S. Reliable assessment of the effects of treatment on mortality and major morbidity, I: clinical trials. Lancet 2001;357:373-80.
308. Schulz KF. Randomised trials, human nature, and reporting guidelines. Lancet 1996;348:596-8.
309. Murray GD. Promoting good research practice. Stat Methods Med Res 2000;9:17-24.
310. Narahari SR, Ryan TJ, Aggithaya MG, Bose KS, Prasanna KS. Evidence-based approaches for the Ayurvedic traditional herbal formulations: toward an Ayurvedic CONSORT model. J Altern Complement Med 2008;14:769-76.
311. Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, Irwig LM, et al. Towards complete and accurate reporting of studies of diagnostic accuracy: The STARD Initiative. Ann Intern Med 2003;138:40-4.
312. von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Ann Intern Med 2007;147:573-7.
313. Tonino PA, De Bruyne B, Pijls NH, Siebert U, Ikeno F, van’t Veer M, et al. Fractional flow reserve versus angiography for guiding percutaneous coronary intervention. N Engl J Med 2009;360:213-24.
Indirizzo per la corrispondenza
dmoher@ohri.caProvenienza
Tradotto con permesso da: Moher D, Hopewell S, Schulz KF, Montori V, Gøtzsche PC, Devereaux PJ, Elbourne D, Egger M, Altman DG. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340:c869.
Membri del CONSORT group che hanno contribuito al CONSORT 2010
Douglas G Altman, Centre for Statistics in Medicine, University of Oxford, UK; Virginia Barbour, PLoS Medicine, UK; Jesse A Berlin, Johnson & Johnson Pharmaceutical Research and Development, USA; Isabelle Boutron, University Paris 7 Denis Diderot, Assistance Publique des Hôpitaux de Paris, INSERM, France; PJ Devereaux, McMaster University, Canada; Kay Dickersin, Johns Hopkins Bloomberg School of Public Health, USA; Diana Elbourne, London School of Hygiene & Tropical Medicine, UK; Susan Ellenberg, University of Pennsylvania School of Medicine, USA; Val Gebski, University of Sydney, Australia; Steven Goodman, Journal of the Society for Clinical Trials, USA; Peter C Gøtzsche, Nordic Cochrane Centre, Denmark; Trish Groves, BMJ, UK; Steven Grunberg, American Society of Clinical Oncology, USA; Brian Haynes, McMaster University, Canada; Sally Hopewell, Centre for Statistics in Medicine, University of Oxford, UK; Astrid James, Lancet; Peter Juhn, Johnson & Johnson, USA; Philippa Middleton, University of Adelaide, Australia; Don Minckler, University of California Irvine, USA; David Moher, Ottawa Methods Centre, Clinical Epidemiology Program, Ottawa Hospital Research Institute, Canada; Victor M Montori, Knowledge and Encounter Research Unit, Mayo Clinic College of Medicine, USA; Cynthia Mulrow, Annals of Internal Medicine, USA; Stuart Pocock, London School of Hygiene & Tropical Medicine, UK; Drummond Rennie, JAMA, USA; David L Schriger, Annals of Emergency Medicine, USA; Kenneth F Schulz, Family Health International, USA; Iveta Simera, EQUATOR Network, UK; Elizabeth Wager, Sideview, UK.
Fonti di finanziamento
Per i finanziamenti ringraziamo: United Kingdom National Institute for Health Research; Canadian Institutes of Health Research; Presidents Fund, Canadian Institutes of Health Research; Johnson & Johnson; BMJ; American Society for Clinical Oncology.
Ringraziamenti
Ringraziamo Frank Davidoff e Tom Lang per la loro partecipazione al CONSORT 2001 Explanation and Elaboration. Un ringraziamento speciale a Maria Ocampo, coordinatore del CONSORT di Ottawa, che ha contribuito a diffondere il CONSORT 2001 Explanation and Elaboration e il CONSORT Statement.
Note alla versione italiana
La Fondazione GIMBE ha sostenuto la traduzione italiana dell’articolo senza alcun supporto istituzionale o commerciale.
Team che ha realizzato la versione italiana
Responsabile scientifico:
Dott. Antonino Cartabellotta
Coordinamento editoriale:
Dott. Marco Mosti
Traduzione:
Dott.ssa Luigia Atorino, Dott.ssa Elena Cottafava
Revisione editoriale:
Dott. Roberto Luceri,
Dott.ssa Francesca Torre
Download
Pagina aggiornata il 27/novembre/2012
Via Amendola, 2 - 40121 Bologna (Italy)
Tel: + 39 051 5883920 - Fax: +39 051 3372195 - info@gimbe.org
CF / P. IVA 03043421209